Separation of Glycyrrhizic Acid and Its Derivants from Hydrolyzation in Subcritical Water by Macroporous Resin

Abstract
:1. Introduction
2. Results and Discussion
2.1. Static Adsorption
2.1.1. Adsorption Kinetics
2.1.2. Adsorption Isotherms
2.2. Static Desorption
The Effect of Different Eluents on the Desorption Ratio for GA, GAMG, and GL
2.3. The Dynamic Adsorption and Desorption
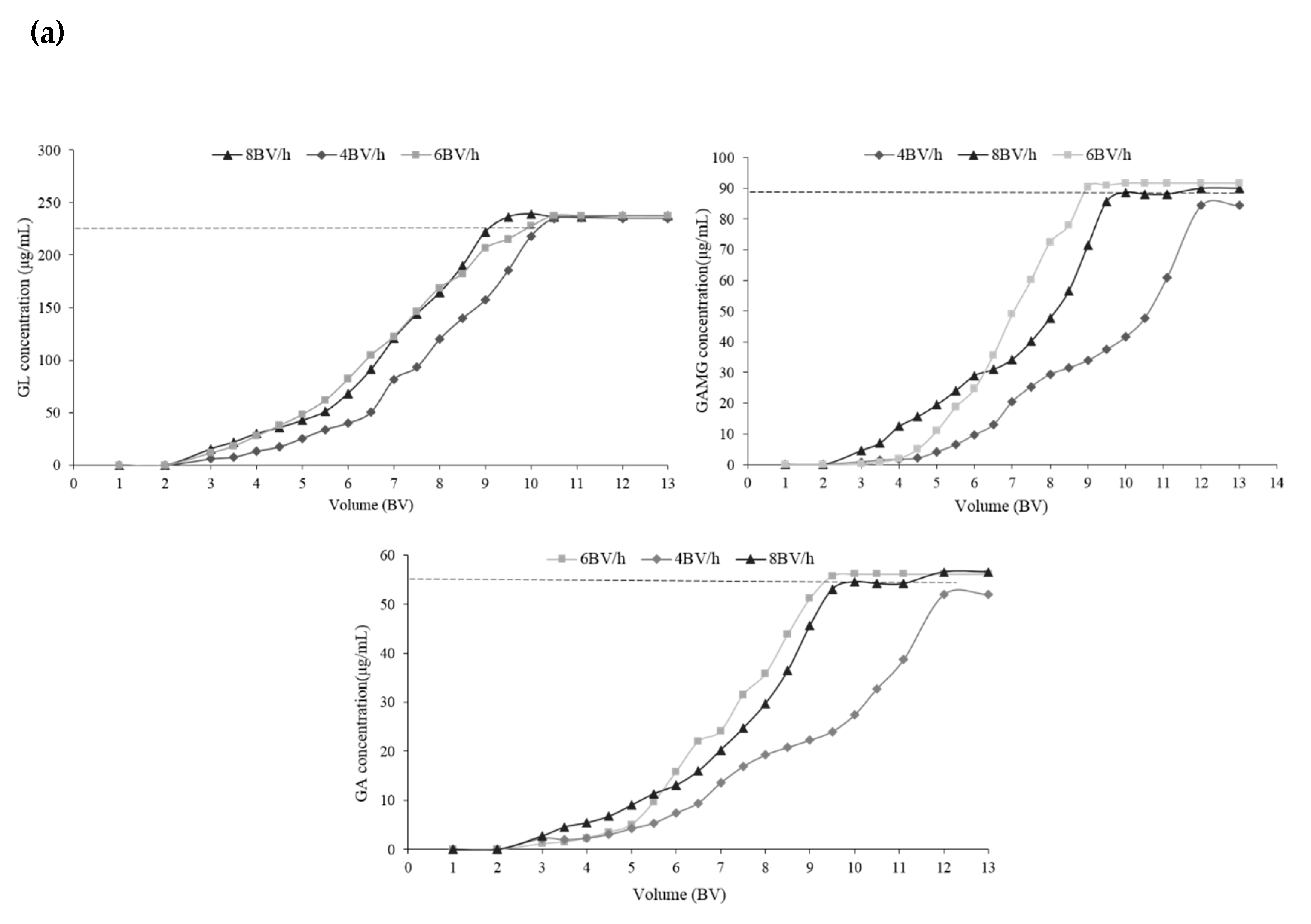
2.3.1. The Dynamic Adsorption Curve
2.3.2. Optimization of the Separation Condition
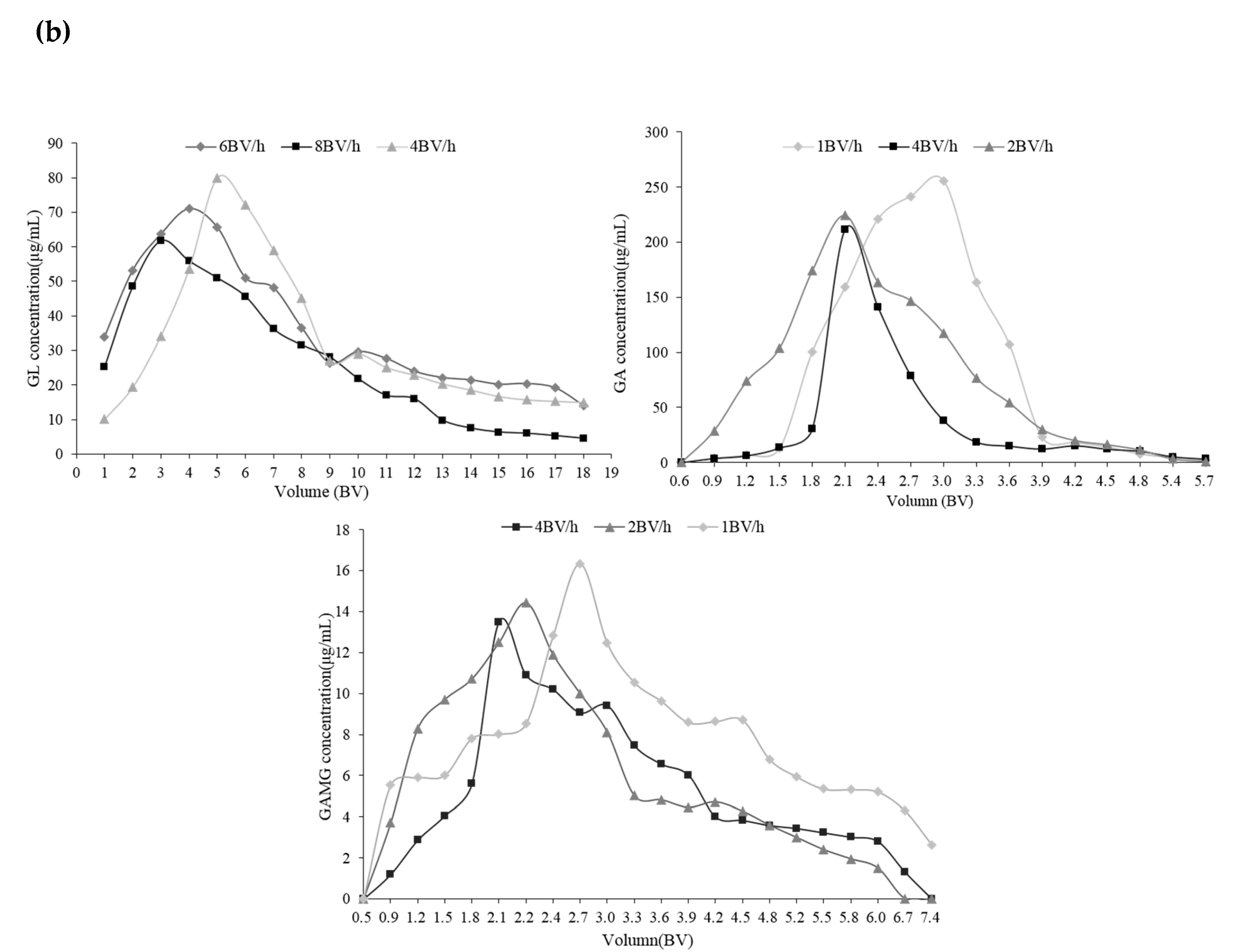
2.3.3. The Multi-Stage Desorption
3. Materials and Methods
3.1. Materials
3.2. Preparation of GL Derivants
3.3. Static Adsorption and Desorption
3.3.1. Static Adsorption
3.3.2. Adsorption Isotherm Modeling
3.3.3. Static Desorption
3.4. Dynamic Adsorption and Desorption
3.4.1. Fixed-Bed Column Adsorption
3.4.2. Optimization of the Desorption Conditions for GL, GAMG, and GA
3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Cinatl, J.; Morgenstern, B.; Bauer, G.; Chandra, P.; Rabenau, H.; Doerr, H. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet 2003, 361, 2045–2046. [Google Scholar] [CrossRef] [Green Version]
- Krähenbühl, S.; Hasler, F.; Krapf, R. Analysis and pharmacokinetics of glycyrrhizic acid and glycyrrhetinic acid in humans and experimental animals. Steroids 1994, 59, 121–126. [Google Scholar] [CrossRef]
- Maatooq, G.T.; Marzouk, A.M.; Gray, A.I.; Rosazza, J.P. Bioactive microbial metabolites from glycyrrhetinic acid. Phytochemistry 2010, 71, 262–270. [Google Scholar] [CrossRef]
- Feng, S.; Li, C.; Xu, X.; Wang, X. Screening strains for directed biosynthesis of β-d-mono-glucuronide-glycyrrhizin and kinetics of enzyme production. J. Mol. Catal. B Enzym. 2006, 43, 63–67. [Google Scholar] [CrossRef]
- Pompei, R.; Flore, O.; Marccialis, M.A.; Pani, A.; Loddo, B. Glycyrrhizic acid inhibits virus growth and inactivates virus particles. Nature 1979, 281, 689–690. [Google Scholar] [CrossRef]
- Baltina, L.A. Chemical modification of glycyrrhizic acid as a route to new bioactive compounds for medicine. Curr. Med. Chem. 2003, 10, 155–171. [Google Scholar] [CrossRef]
- Ohtake, N.; Kido, A.; Kubota, K.; Tsuchiya, N.; Morita, T.; Kase, Y.; Takeda, S. A possible involvement of 3-monoglucuronyl-glycyrrhetinic acid, a metabolite of glycyrrhizin (GL), in GL-induced pseudoaldosteronism. Life Sci. 2007, 80, 1545–1552. [Google Scholar] [CrossRef]
- Huang, H.; Yu, H.; Zhang, J.; Kang, L.; Feng, B.; Song, X.; Ma, B. Biotransformation of glycyrrhizin by Aspergillusniger. Biocatal. Biotransfor. 2009, 27, 90–95. [Google Scholar] [CrossRef]
- Fan, R.; Li, N.; Xu, H.; Xiang, J.; Wang, L.; Gao, Y. The mechanism of hydrothermal hydrolysis for glycyrrhizic acid into glycyrrhetinic acid and glycyrrhetinic acid 3-O-mono-β-d-glucuronide in subcritical water. Food Chem. 2016, 190, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, G.; Curcio, M.; Parisi, O.I.; Puoci, F.; Iemma, F.; Spizzirri, U.; Restuccia, D.; Picci, N. Molecularly imprinted polymers for the selective extraction of glycyrrhizic acid from liquorice roots. Food Chem. 2011, 125, 1058–1063. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Chen, X.; Liu, Y.; Di, D. Preparativeseparation and purification of lycopene from tomato skinsextracts by macroporous adsorption resins. Food Chem. 2010, 123, 1027–1034. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, S.F.; Wu, X.W.; Zhao, X. Macroporous resin adsorption for purification of flavonoids in Houttuyniacordatathunb. Chin. J. Chem. Eng. 2007, 15, 872–876. [Google Scholar]
- Charpe, T.W.; Rathod, V.K. Separation of glycyrrhizic acid from licorice root extract using macroporous resin. Food Bioprod. Process. 2015, 93, 51–57. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Li, A.M.; Sun, J.; Li, P.H. Adsorption of amphoteric aromatic compounds by hyper-cross-linkedresins with amino groups and sulfonic groups. Chem. Eng. J. 2013, 217, 354–362. [Google Scholar] [CrossRef]
- Chang, X.; Xu, H.; Wang, D.; Feng, Y.; Zhan, P. Recovery of Perillaldehyde from hydrodistillation condensate of Perillafrutescens Britton var. crispaf. viridisleaves using macroporous resin adsorption. Sep. Sci. Technol. 2013, 48, 1226–1233. [Google Scholar] [CrossRef]
- He, X.; Dan, J.; Hong, C. Kinetics and Thermodynamics Studies of Glycyrrhizic Acid Adsorption Using S-8 Macroporous Resin. Asian J. Chem. 2013, 25, 9492–9496. [Google Scholar] [CrossRef]
- Tütem, E.; Apak, R.; Ünal, Ç.F. Adsorptive removal of chlorophenols from water by bituminous shale. Water Res. 1998, 32, 2315–2324. [Google Scholar] [CrossRef]
- Barkakati, P.; Begum, A.; Das, M.L.; Rao, P.G. Adsorptive separation of Ginsenoside from aqueous solution by polymeric resins: Equilibrium, kinetic and thermodynamic studies. Chem. Eng. J. 2010, 161, 34–45. [Google Scholar] [CrossRef]
- Gonte, R.R.; Shelar, G.; Balasubramanian, K. Polymer-agro-waste composites for removal of Congo red dye from wastewater: Adsorption isotherms and kinetics. Desalin. Water Treat. 2013, 52, 7797–7811. [Google Scholar] [CrossRef]
- Günay, A.; Arslankaya, E.; Tosun, I. Lead removal from aqueous solution by natural and pretreated clinoptilolite: Adsorption equilibrium and kinetics. J. Hazard. Mater. 2007, 146, 362–371. [Google Scholar] [CrossRef]
- Lorenc, E.; Gryglewicz, G. Adsorption of lignite-derived humic acids on coal-based mesoporous activated carbons. J. Colloid Interface Sci. 2005, 284, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.; McKay, G. Pseudo-second order model for sorption processes. Process. Biochem. 1999, 34, 451–465. [Google Scholar] [CrossRef]
- Liu, Y.; Bai, Q.; Lou, S.; Di, D.; Li, J.; Guo, M. Adsorptioncharacteristics of (−)-epigallocatechingallate and caffeine inthe extract of waste tea on macroporous adsorption resinsfunctionalized with chloromethyl, amino, and phenylaminogroups. J. Agric. Food Chem. 2012, 60, 1555–1566. [Google Scholar] [CrossRef]
- Vasiliu, S.; Bunia, I.; Racovita, S.; Neagu, V. Adsorption of cefotaxime sodium salt on polymer coated ion exchange resin microparticles: Kinetics, equilibrium and thermodynamic studies. Carbohydr. Polym. 2011, 85, 376–387. [Google Scholar] [CrossRef]
- Langmuir, I. The adsorption of gases on plane surfaces of glass, mica and platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef] [Green Version]
- Hall, K.; Eagleton, L.; Acrivos, A.; Vermeulen, T. Pore-andsolid-diffusion kinetics in fixed-bed adsorption underconstant-pattern conditions. Ind. Eng. Chem. Fundam. 1966, 5, 212–223. [Google Scholar] [CrossRef]
- Gao, Z.P.; Yu, Z.F.; Yue, T.L.; Quek, S.Y. Adsorption isotherm, thermodynamics and kinetics studies of polyphenols separation from kiwifruit juice using adsorbent resin. J. Food Eng. 2013, 116, 195–201. [Google Scholar] [CrossRef]
- Wang, T.; Lu, S.; Xia, Q.; Fang, Z.; Johnson, S.K. Separation and purification of amygdalin from thinned bayberry kernels by macroporous adsorption resins. J. Chromatogr. B 2015, 975, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Fu, B.; Liu, J.; Li, H.; Li, L.; Lee, F.S.; Wang, X. The application of macroporous resins in the separation of licorice flavonoids and glycyrrhizic acid. J. Chromatogr. A 2005, 1089, 18–24. [Google Scholar] [CrossRef]
- Bering, B.; Dubinin, M.; Serpinsky, V. On thermodynamics of adsorption in micropores. J. Colloid Interface Sci. 1972, 38, 185–194. [Google Scholar] [CrossRef]
- Liu, Q.R.; Gao, Y.X. Binary adsorption isotherm and kinetics on debittering process of ponkan (Citrus reticulata Blanco) juice with macroporous resins. LWT-Food Sci. Technol. 2015, 63, 1245–1253. [Google Scholar] [CrossRef]
- Lu, D.Q.; Zhang, S.; Li, H.; Dai, Y. Separation of glycyrrhetic acid monoglucuronide from glycyrrhizin hydrolysates by macroporous resins. J. Biotechnol. 2008, 136, 472–473. [Google Scholar] [CrossRef]
- Li, Y.; Cao, R.F.; Wu, X.F.; Huang, J.H.; Deng, S.G.; Lu, X.Y. Hypercrosslinked poly(styrene-co-divinylbenzene) resin as aspecificpolymeric adsorbent for purificationofberberine hydrochloride from aqueous solutions. J. Colloid Interface Sci. 2013, 400, 78–87. [Google Scholar] [CrossRef]
- Niu, G.; Xie, Y.C.; Lou, J.F.; Liu, H. Isolation and purification of glycyrrhizic acid with solvent extraction. Sep. Purif. Technol. 2005, 44, 189–196. [Google Scholar] [CrossRef]
- Zheng, Y.; Wei, J.H.; Qi, L.W.; Cheng, J.M.; Peng, G.P. A green and efficient protocol for large-scale production of glycyrrhizic acid from licorice roots by combination of polyamide and macroporous resin adsorbent chromatography. J. Sep. Sci. 2013, 36, 809–816. [Google Scholar] [CrossRef]
- Wołowicz, A.; Hubicki, Z. Selective Adsorption of Palladium (II) Complexes onto the Chelating Ion Exchange Resin Dowex M 4195-Kinetic Studies. Solvent Extr. Ion. Exch. 2010, 28, 124–159. [Google Scholar] [CrossRef]
- Hayton, J.C.; Allen, D.G.; Scarpello, V. Factor Retention Decisions in Exploratory Factor Analysis: A Tutorial on Parallel Analysis. Organ. Res. Methods 2004, 7, 191–205. [Google Scholar] [CrossRef]
- Rajoriya, R.K.; Prasad, B.; Mishra, I.M.; Wasewar, K.L. Adsorption of benzaldehyde on granular activated carbon: Kinetics, equilibrium, and thermodynamic. Chem. Biochem. Eng. Q. 2007, 21, 219–226. [Google Scholar]
- Chien, S.H.; Clayton, W.R. Application of Elovich Equation to the Kinetics of Phosphate Release and Sorption in Soils. Soil Sci. Soc. Am. J. 1980, 44, 265–268. [Google Scholar] [CrossRef]
- Aharoni, C.; Sideman, S.; Hoffer, E. Adsorption of phosphate ions by collodion-coated alumina. J. Chem. Technol. Biotechnol. 2007, 29, 404–412. [Google Scholar] [CrossRef]
- Rahmani, A.; Hosseini, A.; Hosseini, S.; Kharghani, K.; Zarei, H.; Rastegar, A. Kinetic, equilibrium and thermodynamic studies on sorption of uranium and thorium from aqueous solutions by a selective impregnated resin containing carminic acid. J. Hazard. Mater. 2015, 286, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.; Pompeu, D.; Larondelle, Y.; Rogez, H. Optimisation of the adsorption of polyphenols from Inga edulis leaves on macroporous resins using an experimental design methodology. Sep. Purif. Technol. 2007, 53, 274–280. [Google Scholar] [CrossRef]
- Dubinin, M.M.; Radushkevich, L.V. Equation of the characteristic curve of activated charcoal. Chem. Zentr. 1947, 1, 875. [Google Scholar]
- Zhou, L.; Shang, C.; Liu, Z.; Huang, G.; Adesina, A.A. Selective adsorption of uranium (VI) from aqueous solutions using the ion-imprinted magnetic chitosan resins. J. Colloid Interface Sci. 2012, 366, 165–172. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Kinetic Parameters for Adsorption of GL, GAMG, and GA | ||||||
|---|---|---|---|---|---|---|
| Model | Parameters | |||||
| Pseudo first-order | k1 | Qe,cal(mg/g) | Qe,exp(mg/g) | R2 | Q(%) | Af |
| GL | 0.066 | 8.661 | 10.888 | 0.815 | 18.05 | 1.458 |
| GAMG | 0.122 | 2.948 | 2.638 | 0.890 | 15.89 | 1.182 |
| GA | 0.116 | 3.081 | 2.468 | 0.845 | 19.05 | 1.294 |
| Pseudo second-order | k2 | Qe,cal(mg/g) | Qe,exp(mg/g) | R2 | Q(%) | Af |
| GL | 0.069 | 10.225 | 10.888 | 0.999 | 7.20 | 1.042 |
| GAMG | 0.113 | 2.880 | 2.638 | 0.986 | 10.68 | 1.089 |
| GA | 0.094 | 2.744 | 2.468 | 0.980 | 9.25 | 1.104 |
| Intra-particle diffusion | Kid[g/(g·h1/2)] | C | R2 | Q(%) | Af | |
| GL | 1.147 | 4.892 | 0.862 | 7.012 | 1.0587 | |
| GAMG | 0.446 | 0.663 | 0.810 | 14.38 | 1.1083 | |
| GA | 0.450 | 0.430 | 0.857 | 10.518 | 1.0896 | |
| Boyd’s diffusion | De(m2/s) | R2 | Q(%) | Af | ||
| GL | 7.7 × 10−6 | 0.838 | 16.068 | 1.1685 | ||
| GAMG | 1.1 × 10−5 | 0.901 | 11.758 | 1.1035 | ||
| GA | 9.4 × 10−6 | 0.847 | 13.051 | 1.1026 | ||
| Elovich diffusion | α[mg/(g·min)] | β(g/mg) | R2 | Q(%) | Af | |
| GL | 0.724 | 0.642 | 0.940 | 4.54 | 1.037 | |
| GAMG | 0.037 | 1.677 | 0.890 | 12.76 | 1.097 | |
| GA | 0.027 | 1.707 | 0.925 | 11.02 | 1.090 | |
| Bangham diffusion | α | k0((mL/(g·L)) | R2 | Q(%) | Af | |
| GL | 0.260 | 0.053 | 0.936 | 4.61 | 1.039 | |
| GAMG | 0.460 | 0.016 | 0.886 | 11.47 | 1.097 | |
| GA | 0.541 | 0.013 | 0.943 | 8.45 | 1.067 | |
| Model | Parameters | |||||
|---|---|---|---|---|---|---|
| Langmuir | Q0 (mg/g) | aL (L/mg) | RL | KL (L/g) | R2 | RMSE |
| 34.722 | 0.009 | 0.068 | 0.328 | 0.988 | 1.104 | |
| Freundlich | 1/n | KF (mg/g) | R2 | RMSE | ||
| 0.719 | 0.482 | 0.993 | 0.974 | |||
| Temkin | bT (J/mol) | KT (L/mg) | −∆G (kJ/mol) | R2 | RMSE | |
| 7.809 | 0.165 | 4.384 | 0.842 | 1.303 | ||
| D-R | Q0 (mg/g) | ε (kJ/mol) | β (10−5, (mol2/J2) | R2 | RMSE | |
| 25.977 | 0.224 | 2.000 | 0.888 | 1.121 | ||
| Model | Adsorbate | Parameters | |||||
|---|---|---|---|---|---|---|---|
| Langmuir | Q0 (mg/g) | aL (L/mg) | KL (L/g) | RL | R2 | RMSE | |
| GL | 13.175 | 0.573 | 7.550 | 0.005 | 0.853 | 1.223 | |
| GAMG | 3.211 | 1.843 | 5.919 | 0.009 | 0.859 | 1.567 | |
| GA | 2.276 | 2.871 | 6.534 | 0.008 | 0.889 | 1.078 | |
| Freundlich | 1/n | KF (mg/g) | R2 | RMSE | |||
| GL | 0.4162 | 0.956 | 0.824 | 2.012 | |||
| GAMG | 0.471 | 0.384 | 0.882 | 1.712 | |||
| GA | 0.505 | 0.281 | 0.922 | 0.989 | |||
| Temkin | bT (J/mol) | KT (L/mg) | −∆G (KJ/mol) | R2 | RMSE | ||
| GL | 3.004 | 0.092 | 5.812 | 0.829 | 2.145 | ||
| GAMG | 0.749 | 0.453 | 1.930 | 0.903 | 1.567 | ||
| GA | 0.567 | 0.509 | 1.643 | 0.937 | 0.898 | ||
| D-R | Q0 (mg/g) | β (10−5, mol2/J2) | ε (KJ/mol) | R2 | RMSE | ||
| GL | 9.456 | 50.000 | 0.032 | 0.854 | 1.898 | ||
| GAMG | 2.173 | 1.000 | 0.224 | 0.880 | 1.679 | ||
| GA | 1.498 | 0.070 | 0.267 | 0.824 | 2.123 | ||
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, R. Separation of Glycyrrhizic Acid and Its Derivants from Hydrolyzation in Subcritical Water by Macroporous Resin. Molecules 2020, 25, 4305. https://doi.org/10.3390/molecules25184305
Fan R. Separation of Glycyrrhizic Acid and Its Derivants from Hydrolyzation in Subcritical Water by Macroporous Resin. Molecules. 2020; 25(18):4305. https://doi.org/10.3390/molecules25184305
Chicago/Turabian StyleFan, Rui. 2020. "Separation of Glycyrrhizic Acid and Its Derivants from Hydrolyzation in Subcritical Water by Macroporous Resin" Molecules 25, no. 18: 4305. https://doi.org/10.3390/molecules25184305
APA StyleFan, R. (2020). Separation of Glycyrrhizic Acid and Its Derivants from Hydrolyzation in Subcritical Water by Macroporous Resin. Molecules, 25(18), 4305. https://doi.org/10.3390/molecules25184305