
Considerations for Docking of Selective Angiotensin-Converting Enzyme Inhibitors


Abstract
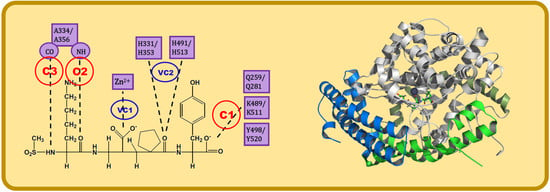
:
1. Introduction
2. Rational Design of ACE Inhibitors and Crystallographic Structural Information
2.1. Structure of Both ACE Domains
2.2. Molecular Modeling Applied to the Study of ACE Inhibitors in the Last Years
2.3. Molecular Modeling for the Development of Novel Selective Inhibitors
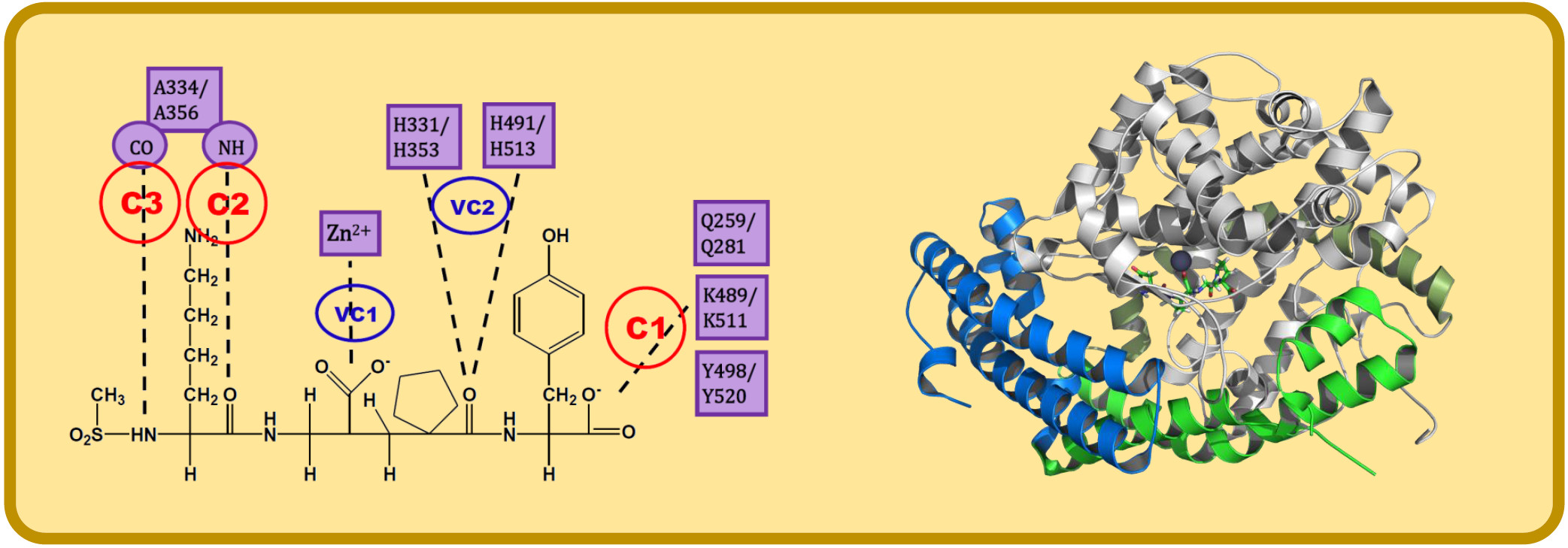
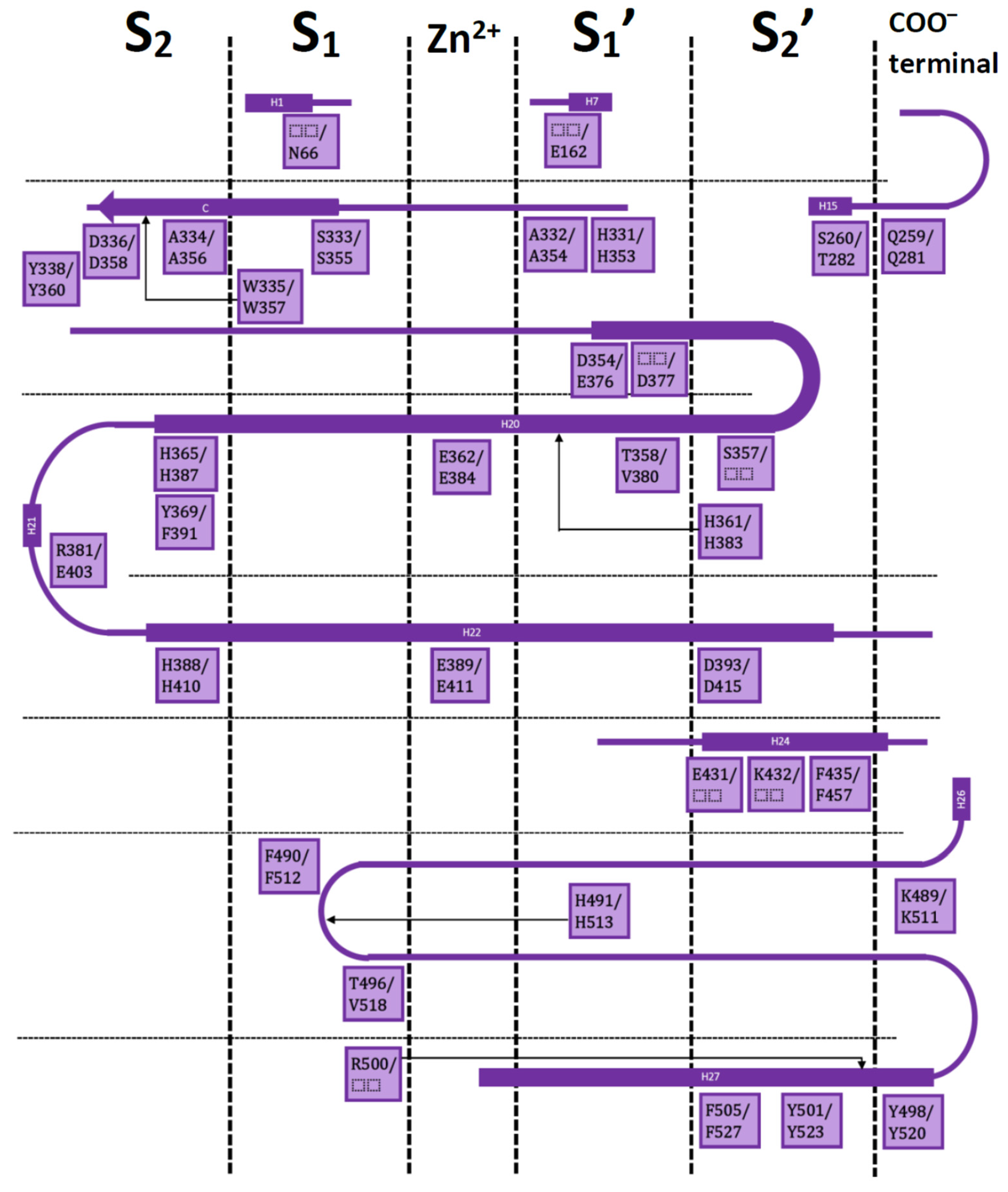
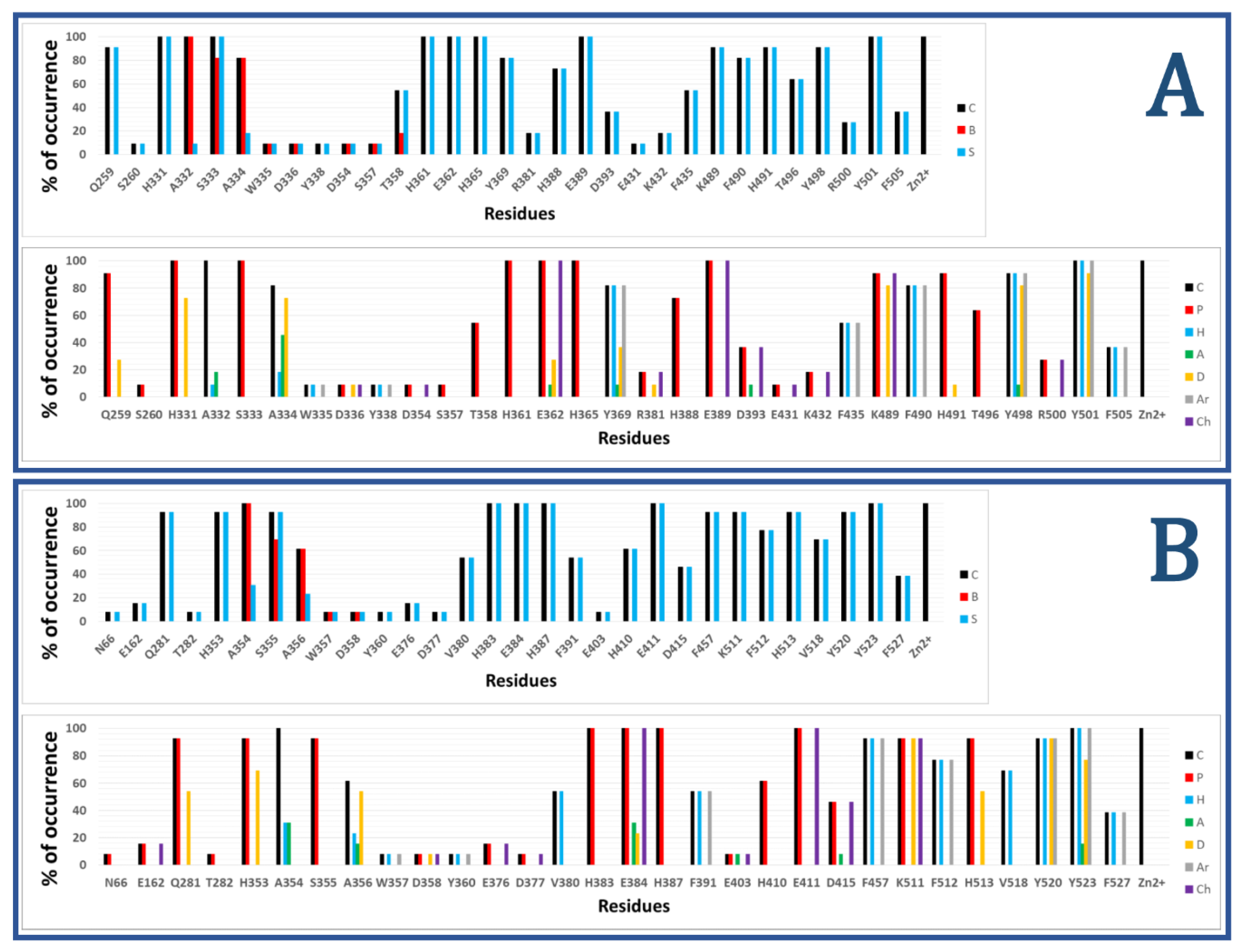
3. Interaction Fingerprints (IFPs) for ACE Inhibitors Derived from PDB Structures
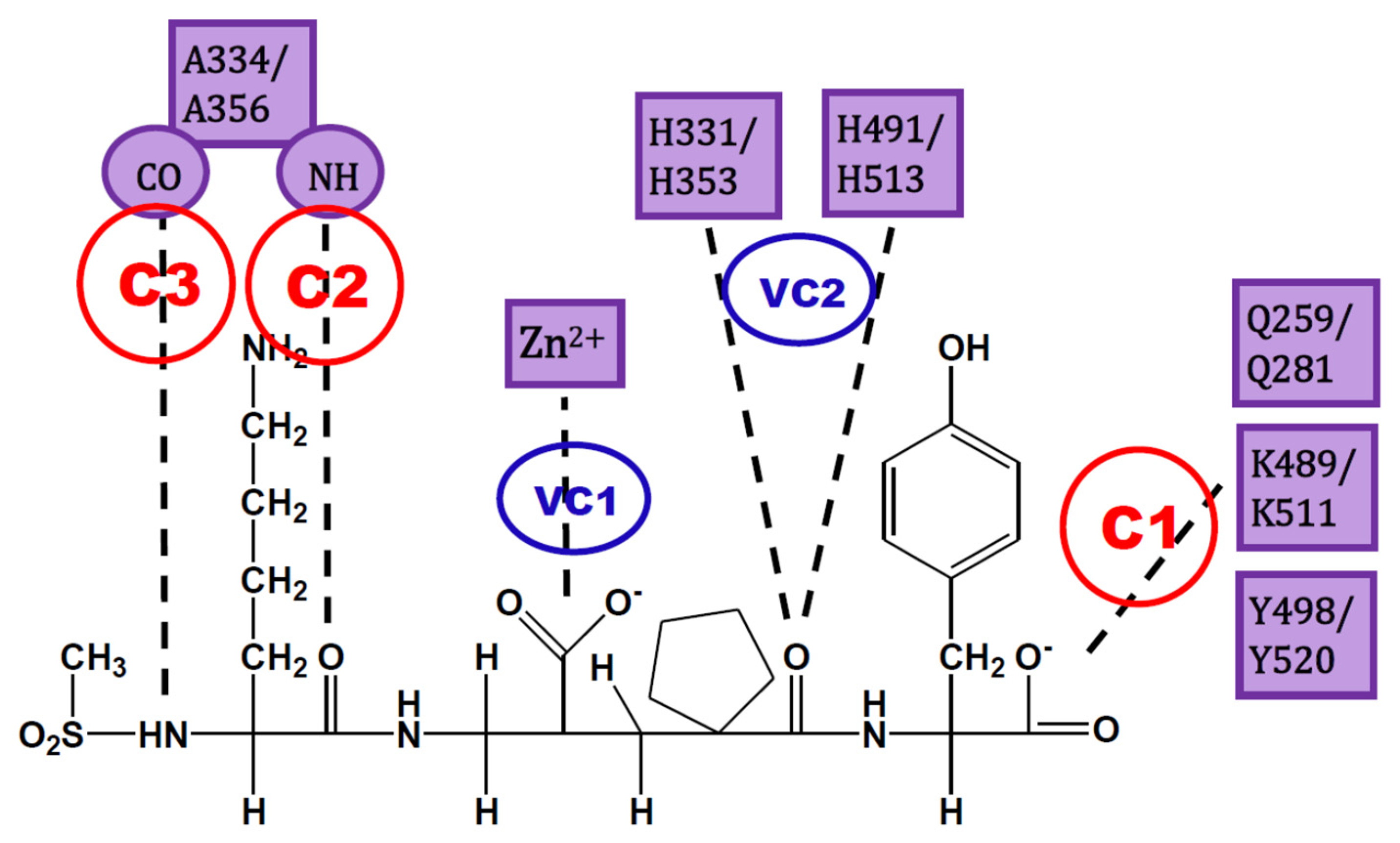
4. Docking of Selective ACE Inhibitors is Facilitated by the Use of Constraints
- C1:
- A C-terminal group of the ligand must be an HB acceptor to K489/K511 from nACE/cACE and it must have proximity to Q259/Q281 and Y498/Y520 from nACE/cACE.
- VC1:
- The ligand must have an HB acceptor group at P1′ to form HB with H331 and/or H491 (nACE) or H353 and/or H513 (cACE). Both histidine residues in nACE and cACE must have a proton at position ε (this consideration should be important when molecular mechanics methods are used).
- VC2:
- The ligand must have a polar group (a negatively charged group or an electron donor group) forming interactions with the Zn2+ ion.
- C2:
- Ligand must be an HB acceptor to NH of Ala334 (nACE) or Ala356 (cACE)
- C3:
- Ligand must be an HB donor to CO of Ala334 (nACE) or Ala356 (cACE).
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Soffer, R.L. Angiotensin-converting enzyme and the regulation of vasoactive peptides. Annu. Rev. Biochem. 1976, 45, 73–94. [Google Scholar] [CrossRef] [PubMed]
- Ondetti, M.A.; Rubin, B.; Cushman, D.W. Design of specific inhibitors of angiotensin-converting enzyme: New class of orally active antihypertensive agents. Science 1977, 196, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Cushman, D.W.; Cheung, H.S.; Sabo, E.F.; Ondetti, M.A. Design of potent competitive inhibitors of angiotensin-converting enzyme. Carboxyalkanoyl and mercaptoalkanoyl amino acids. Biochemistry 1977, 16, 5484–5491. [Google Scholar] [CrossRef] [PubMed]
- Opie, L.H.; Kowolik, H. The discovery of captopril: From large animals to small molecules. Cardiovasc. Res. 1995, 30, 18–25. [Google Scholar] [CrossRef]
- Ferreira, S.H.; Bartelt, D.C.; Greene, L.J. Isolation of bradykinin-potentiating peptides from Bothrops jararaca venom. Biochemistry 1970, 9, 2583–2593. [Google Scholar] [CrossRef]
- Cushman, D.W.; Pluscec, J.; Williams, N.J.; Weaver, E.R.; Sabo, E.F.; Kocy, O.; Cheung, H.S.; Ondetti, M.A. Inhibition of angiotensin-coverting enzyme by analogs of peptides from Bothrops jararaca venom. Experientia 1973, 29, 1032–1035. [Google Scholar] [CrossRef]
- Ulm, E.H.; Hichens, M.; Gomez, H.J.; Till, A.E.; Hand, E.; Vassil, T.C.; Biollaz, J.; Brunner, H.R.; Schelling, J.L. Enalapril maleate and a lysine analogue (MK-521): Disposition in man. Br. J. Clin. Pharmacol. 1982, 14, 357–362. [Google Scholar] [CrossRef] [Green Version]
- Song, J.C.; White, C.M. Clinical pharmacokinetics and selective pharmacodynamics of new angiotensin converting enzyme inhibitors: An update. Clin Pharm. 2002, 41, 207–224. [Google Scholar] [CrossRef]
- Nathisuwan, S.; Talbert, R.L. A review of vasopeptidase inhibitors: A new modality in the treatment of hypertension and chronic heart failure. Pharmacotherapy 2002, 22, 27–42. [Google Scholar] [CrossRef]
- Nussberger, J.; Cugno, M.; Amstutz, C.; Cicardi, M.; Pellacani, A.; Agostoni, A. Plasma bradykinin in angio-oedema. The Lancet 1998, 351, 1693–1697. [Google Scholar] [CrossRef]
- Bernstein, K.E.; Shen, X.Z.; Gonzalez-Villalobos, R.A.; Billet, S.; Okwan-Duodu, D.; Ong, F.S.; Fuchs, S. Different in vivo functions of the two catalytic domains of angiotensin-converting enzyme (ACE). Curr. Opin. Pharmacol. 2011, 11, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, S.; Xiao, H.D.; Hubert, C.; Michaud, A.; Campbell, D.J.; Adams, J.W.; Capecchi, M.R.; Corvol, P.; Bernstein, K.E. Angiotensin-converting enzyme C-terminal catalytic domain is the main site of angiotensin I cleavage in vivo. Hypertension 2008, 51, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Yin, C. The anti-inflammatory peptide Ac-SDKP: Synthesis, role in ACE inhibition, and its therapeutic potential in hypertension and cardiovascular diseases. Pharmacol. Res. 2018, 134, 268–279. [Google Scholar] [CrossRef]
- Messerli, F.H.; Nussberger, J. Vasopeptidase inhibition and angio-oedema. The Lancet 2000, 356, 608–609. [Google Scholar] [CrossRef]
- Cotton, J.; Hayashi, M.A.F.; Cuniasse, P.; Vazeux, G.; Ianzer, D.; De Camargo, A.C.M.; Dive, V. Selective inhibition of the C-domain of angiotensin I converting enzyme by bradykinin potentiating peptides. Biochemistry 2002, 41, 6065–6071. [Google Scholar] [CrossRef]
- Sharma, U.; Rhaleb, N.-E.; Pokharel, S.; Harding, P.; Rasoul, S.; Peng, H.; Carretero, O.A. Novel anti-inflammatory mechanisms of N-Acetyl-Ser-Asp-Lys-Pro in hypertension-induced target organ damage. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1226–H1232. [Google Scholar] [CrossRef]
- Acharya, K.R.; Sturrock, E.D.; Riordan, J.F.; Ehlers, M.R.W. Ace revisited: A new target for structure-based drug design. Nat. Rev. Drug Discov. 2003, 2, 891–902. [Google Scholar] [CrossRef]
- Dive, V.; Cotton, J.; Yiotakis, A.; Michaud, A.; Vassiliou, S.; Jiracek, J.; Vazeux, G.; Chauvet, M.T.; Cuniasse, P.; Corvol, P. RXP 407, a phosphinic peptide, is a potent inhibitor of angiotensin I converting enzyme able to differentiate between its two active sites. Proc. Natl. Acad. Sci. USA 1999, 96, 4330–4335. [Google Scholar] [CrossRef] [Green Version]
- Georgiadis, D.; Cuniasse, P.; Cotton, J.; Yiotakis, A.; Dive, V. Structural determinants of RXPA380, a potent and highly selective inhibitor of the angiotensin-converting enzyme C-domain. Biochemistry 2004, 43, 8048–8054. [Google Scholar] [CrossRef]
- Anthony, C.S.; Corradi, H.R.; Schwager, S.L.U.; Redelinghuys, P.; Georgiadis, D.; Dive, V.; Acharya, K.R.; Sturrock, E.D. The N domain of human angiotensin-I-converting enzyme: The role of N-glycosylation and the crystal structure in complex with an N domain-specific phosphinic inhibitor, RXP407. J. Biol. Chem. 2010, 285, 35685–35693. [Google Scholar] [CrossRef] [Green Version]
- Ashok, A.; Brijesha, N.; Aparna, H.S. Discovery, synthesis, and in vitro evaluation of a novel bioactive peptide for ACE and DPP-IV inhibitory activity. Eur. J. Med. Chem. 2019, 180, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Ugwu, C.P.; Abarshi, M.M.; Mada, S.B.; Sanusi, B.; Nzelibe, H.C. Camel and horse milk casein hydrolysates exhibit angiotensin converting enzyme inhibitory and antioxidative effects in vitro and in silico. Int. J. Pept. Res. Ther. 2019, 25, 1595–1604. [Google Scholar] [CrossRef]
- Xie, J.; Chen, X.; Wu, J.; Zhang, Y.; Zhou, Y.; Zhang, L.; Tang, Y.-J.; Wei, D. Antihypertensive effects, molecular docking study, and isothermal titration calorimetry assay of angiotensin I-converting enzyme inhibitory peptides from chlorella vulgaris. J. Agric. Food Chem. 2018, 66, 1359–1368. [Google Scholar] [CrossRef]
- Liu, L.; Wei, Y.; Chang, Q.; Sun, H.; Chai, K.; Huang, Z.; Zhao, Z.; Zhao, Z. Ultrafast Screening of a Novel, Moderately Hydrophilic Angiotensin-Converting-Enzyme-Inhibitory Peptide, RYL, from Silkworm Pupa Using an Fe-Doped-Silkworm-Excrement-Derived Biocarbon: Waste Conversion by Waste. J. Agric. Food Chem. 2017, 65, 11202–11211. [Google Scholar] [CrossRef] [PubMed]
- Priyanto, A.D.; Doerksen, R.J.; Chang, C.-I.; Sung, W.-C.; Widjanarko, S.B.; Kusnadi, J.; Lin, Y.-C.; Wang, T.-C.; Hsu, J.-L. Screening, discovery, and characterization of angiotensin-I converting enzyme inhibitory peptides derived from proteolytic hydrolysate of bitter melon seed proteins. J. Proteom. 2015, 128, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Baby, B.; Soman, S.S.; Vijayan, R. Molecular insights into the interaction of hemorphin and its targets. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Li, Y.; Peng, K.; Wang, X.-L.; Ding, Z.; Liu, L.; Xu, P.; Liu, G.-Q. Isolation and Characterization of Three Antihypertension Peptides from the Mycelia of Ganoderma Lucidum (Agaricomycetes). J. Agric. Food Chem. 2019, 67, 8149–8159. [Google Scholar] [CrossRef]
- Gao, X.; Li, X.; Yan, P.; Sun, R.; Kan, G.; Zhou, Y. Identification and Functional Mechanism of Novel Angiotensin I Converting Enzyme Inhibitory Dipeptides from Xerocomus badius Cultured in Shrimp Processing Waste Medium. Biomed Res. Int. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Taga, Y.; Hayashida, O.; Ashour, A.; Amen, Y.; Kusubata, M.; Ogawa-Goto, K.; Shimizu, K.; Hattori, S. Characterization of Angiotensin-Converting Enzyme Inhibitory Activity of X-Hyp-Gly-Type Tripeptides: Importance of Collagen-Specific Prolyl Hydroxylation. J. Agric. Food Chem. 2018, 66, 8737–8743. [Google Scholar] [CrossRef]
- Yu, Z.; Fan, Y.; Zhao, W.; Ding, L.; Li, J.; Liu, J. Novel Angiotensin-Converting Enzyme Inhibitory Peptides Derived from Oncorhynchus mykiss Nebulin: Virtual Screening and In Silico Molecular Docking Study. J. Food Sci. 2018, 83, 2375–2383. [Google Scholar] [CrossRef]
- Yu, D.; Wang, C.; Song, Y.; Zhu, J.; Zhang, X. Discovery of Novel Angiotensin-Converting Enzyme Inhibitory Peptides from Todarodes pacificus and Their Inhibitory Mechanism: In Silico and In Vitro Studies. Int. J. Mol. Sci. 2019, 20, 4159. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.; Zhang, L.; Han, X.; Meng, Z.; Zhang, J.; Wu, Y.; Cheng, D. Quantitative Structure-Activity Relationship Modeling Coupled with Molecular Docking Analysis in Screening of Angiotensin I-Converting Enzyme Inhibitory Peptides from Qula Casein Hydrolysates Obtained by Two-Enzyme Combination Hydrolysis. J. Agric. Food Chem. 2018, 66, 3221–3228. [Google Scholar] [CrossRef]
- Li, J.; Liu, Z.; Zhao, Y.; Zhu, X.; Yu, R.; Dong, S.; Wu, H. Novel Natural Angiotensin Converting Enzyme (ACE)-Inhibitory Peptides Derived from Sea Cucumber-Modified Hydrolysates by Adding Exogenous Proline and a Study of Their Structure–Activity Relationship. Mar. Drugs 2018, 16, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, W.; Chen, C.; Zeng, H.; Lin, J.; Zhang, Y.; Hu, J.; Zheng, B. Novel angiotensin-converting enzyme inhibitory peptides derived from Trichiurus lepturus myosin: Molecular docking and surface plasmon resonance study. LWT 2019, 110, 54–63. [Google Scholar] [CrossRef]
- Abdelhedi, O.; Nasri, R.; Mora, L.; Jridi, M.; Toldrá, F.; Nasri, M. In silico analysis and molecular docking study of angiotensin I-converting enzyme inhibitory peptides from smooth-hound viscera protein hydrolysates fractionated by ultrafiltration. Food Chem. 2018, 239, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.; Mirdamadi, S.; Ehsani, M.R.; Aminlari, M. Production of antioxidant and ACE-inhibitory peptides from Kluyveromyces marxianus protein hydrolysates: Purification and molecular docking. J. Food Drug Anal. 2018, 26, 696–705. [Google Scholar] [CrossRef] [Green Version]
- Tu, M.; Wang, C.; Chen, C.; Zhang, R.; Liu, H.; Lu, W.; Jiang, L.; Du, M. Identification of a novel ACE-inhibitory peptide from casein and evaluation of the inhibitory mechanisms. Food Chem. 2018, 256, 98–104. [Google Scholar] [CrossRef]
- Amorim, F.G.; Coitinho, L.B.; Dias, A.T.; Friques, A.G.F.; Monteiro, B.L.; de Rezende, L.C.D.; de Melo Costa Pereira, T.; Campagnaro, B.P.; de Pauw, E.; Vasquez, E.C.; et al. Identification of new bioactive peptides from Kefir milk through proteopeptidomics: Bioprospection of antihypertensive molecules. Food Chem. 2019, 282, 109–119. [Google Scholar] [CrossRef]
- Nawaz, K.A.A.; David, S.M.; Murugesh, E.; Thandeeswaran, M.; Kiran, K.G.; Mahendran, R.; Palaniswamy, M.; Angayarkanni, J. Identification and in silico characterization of a novel peptide inhibitor of angiotensin converting enzyme from pigeon pea (Cajanus cajan). Phytomedicine 2017, 36, 1–7. [Google Scholar] [CrossRef]
- Tu, M.; Liu, H.; Zhang, R.; Chen, H.; Mao, F.; Cheng, S.; Lu, W.; Du, M. Analysis and Evaluation of the Inhibitory Mechanism of a Novel Angiotensin-I-Converting Enzyme Inhibitory Peptide Derived from Casein Hydrolysate. J. Agric. Food Chem. 2018, 66, 4139–4144. [Google Scholar] [CrossRef]
- Assaran Darban, R.; Shareghi, B.; Asoodeh, A.; Chamani, J. Multi-spectroscopic and molecular modeling studies of interaction between two different angiotensin I converting enzyme inhibitory peptides from gluten hydrolysate and human serum albumin. J. Biomol. Struct. Dyn. 2017, 35, 3648–3662. [Google Scholar] [CrossRef] [PubMed]
- Zarei, M.; Abidin, N.B.Z.; Auwal, S.M.; Chay, S.Y.; Haiyee, Z.A.; Sikin, A.M.; Saari, N. Angiotensin Converting Enzyme (ACE)-Peptide Interactions: Inhibition Kinetics, In Silico Molecular Docking and Stability Study of Three Novel Peptides Generated from Palm Kernel Cake Proteins. Biomolecules 2019, 9, 569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, C.; Sun, L.-C.; Yan, L.-J.; Lin, Y.-C.; Liu, G.-M.; Cao, M.-J. Production, optimisation and characterisation of angiotensin converting enzyme inhibitory peptides from sea cucumber (Stichopus japonicus) gonad. Food Funct. 2018, 9, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tu, M.; Wu, D.; Chen, H.; Chen, C.; Wang, Z.; Jiang, L. Identification of an ACE-Inhibitory Peptide from Walnut Protein and Its Evaluation of the Inhibitory Mechanism. Int. J. Mol. Sci. 2018, 19, 1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, Y.; Liping, S.; Yongliang, Z. Preparation and identification of novel inhibitory angiotensin-I-converting enzyme peptides from tilapia skin gelatin hydrolysates: Inhibition kinetics and molecular docking. Food Funct. 2018, 9, 5251–5259. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Zhang, Z.; Luo, L.; Zhu, J.; Huang, F.; Yang, Z.; Tang, Y.; Ding, G. Identification and Molecular Docking Study of a Novel Angiotensin-I Converting Enzyme Inhibitory Peptide Derived from Enzymatic Hydrolysates of Cyclina sinensis. Mar. Drugs 2018, 16, 411. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Xu, X.; Sun, X.; Zhang, X.; Chen, X.; Xu, N. Preparation and Identification of ACE Inhibitory Peptides from the Marine Macroalga Ulva intestinalis. Mar. Drugs 2019, 17, 179. [Google Scholar] [CrossRef] [Green Version]
- Joel, C.H.; Sutopo, C.C.Y.; Prajitno, A.; Su, J.-H.; Hsu, J.-L. Screening of Angiotensin-I Converting Enzyme Inhibitory Peptides Derived from Caulerpa lentillifera. Molecules 2018, 23, 3005. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, Y.; Chen, P.; Shu, F.; Li, K.; Qiao, L.; Chen, Z.; Wang, L. A novel angiotensin-I converting enzyme inhibitory peptide derived from the glutelin of vinegar soaked black soybean and its antihypertensive effect in spontaneously hypertensive rats. J. Biochem. 2019, 166, 223–230. [Google Scholar] [CrossRef]
- Marseglia, A.; Dellafiora, L.; Prandi, B.; Lolli, V.; Sforza, S.; Cozzini, P.; Tedeschi, T.; Galaverna, G.; Caligiani, A. Simulated Gastrointestinal Digestion of Cocoa: Detection of Resistant Peptides and In Silico/In Vitro Prediction of Their Ace Inhibitory Activity. Nutrients 2019, 11, 985. [Google Scholar] [CrossRef] [Green Version]
- Pinciroli, M.; Aphalo, P.; Nardo, A.E.; Añón, M.C.; Quiroga, A.V. Broken Rice as a Potential Functional Ingredient with Inhibitory Activity of Renin and Angiotensin-Converting Enzyme (ACE). Plant Foods Hum. Nutr. 2019, 74, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Setayesh-Mehr, Z.; Asoodeh, A. The inhibitory activity of HL-7 and HL-10 peptide from scorpion venom (Hemiscorpius lepturus) on angiotensin converting enzyme: Kinetic and docking study. Bioorg. Chem. 2017, 75, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Kharazmi-Khorassani, J.; Asoodeh, A.; Tanzadehpanah, H. Antioxidant and angiotensin-converting enzyme (ACE) inhibitory activity of thymosin alpha-1 (Thα1) peptide. Bioorg. Chem. 2019, 87, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Savitha, M.N.; Siddesha, J.M.; Suvilesh, K.N.; Yariswamy, M.; Vivek, H.K.; D’Souza, C.J.M.; Umashankar, M.; Vishwanath, B.S. Active-site directed peptide l-Phe-d-His-l-Leu inhibits angiotensin converting enzyme activity and dexamethasone-induced hypertension in rats. Peptides 2019, 112, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Fang, L.; Min, W.; Liu, J.; Li, H. Exploration of the molecular interactions between angiotensin-I-converting enzyme (ACE) and the inhibitory peptides derived from hazelnut (Corylus heterophylla Fisch.). Food Chem. 2018, 245, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Sun, L.; Muhammad, Y.; Wang, Z.; Liu, H.; Sun, J.; Zhou, L.; Feng, X.; Liao, D.; Wang, S. Studies on the Interaction between Angiotensin-Converting Enzyme (ACE) and ACE Inhibitory Peptide from Saurida elongata. J. Agric. Food Chem. 2018, 66, 13414–13422. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhang, H.; Bian, X.; Li, J.; Li, J.; Zhang, H. Insight into the binding of ACE-inhibitory peptides to angiotensin-converting enzyme: A molecular simulation. Mol. Simul. 2019, 45, 215–222. [Google Scholar] [CrossRef]
- Qi, C.; Lin, G.; Zhang, R.; Wu, W. Studies on the Bioactivities of ACE-inhibitory Peptides with Phenylalanine C-terminus Using 3D-QSAR, Molecular Docking and in vitro Evaluation. Mol. Inform. 2017, 36, 1600157. [Google Scholar] [CrossRef]
- Qi, C.; Zhang, R.; Liu, F.; Zheng, T.; Wu, W. Molecular mechanism of interactions between inhibitory tripeptide GEF and angiotensin-converting enzyme in aqueous solutions by molecular dynamic simulations. J. Mol. Liq. 2018, 249, 389–396. [Google Scholar] [CrossRef]
- Fang, L.; Geng, M.; Liu, C.; Wang, J.; Min, W.; Liu, J. Structural and molecular basis of angiotensin-converting enzyme by computational modeling: Insights into the mechanisms of different inhibitors. PLoS ONE 2019, 14, e0215609. [Google Scholar] [CrossRef]
- Tong, J.; Li, L.; Bai, M.; Li, K. A New Descriptor of Amino Acids-SVGER and its Applications in Peptide QSAR. Mol. Inform. 2017, 36, 1501023. [Google Scholar] [CrossRef] [PubMed]
- Deng, B.; Ni, X.; Zhai, Z.; Tang, T.; Tan, C.; Yan, Y.; Deng, J.; Yin, Y. New Quantitative Structure-Activity Relationship Model for Angiotensin-Converting Enzyme Inhibitory Dipeptides Based on Integrated Descriptors. J. Agric. Food Chem. 2017, 65, 9774–9781. [Google Scholar] [CrossRef] [PubMed]
- Forero, D.P.; Masatani, C.; Fujimoto, Y.; Coy-Barrera, E.; Peterson, D.G.; Osorio, C. Spermidine Derivatives in Lulo (Solanum quitoense Lam.) Fruit: Sensory (Taste) versus Biofunctional (ACE-Inhibition) Properties. J. Agric. Food Chem. 2016, 64, 5375–5383. [Google Scholar] [CrossRef] [PubMed]
- Salehabadi, H.; Khajeh, K.; Dabirmanesh, B.; Biglar, M.; Amanlou, M. Evaluation of angiotensin converting enzyme inhibitors by SPR biosensor and theoretical studies. Enzym. Microb. Technol. 2019, 120, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Maneesh, A.; Chakraborty, K. Previously undescribed antioxidative O-heterocyclic angiotensin converting enzyme inhibitors from the intertidal seaweed Sargassum wightii as potential antihypertensives. Food Res. Int. 2018, 113, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Hai-Bang, T.; Shimizu, K. Structure–activity relationship and inhibition pattern of reishi-derived (Ganoderma lingzhi) triterpenoids against angiotensin-converting enzyme. Phytochem. Lett. 2015, 12, 243–247. [Google Scholar] [CrossRef]
- Ali, M.Y.; Seong, S.H.; Jung, H.A.; Choi, J.S. Angiotensin-I-Converting Enzyme Inhibitory Activity of Coumarins from Angelica decursiva. Molecules 2019, 24, 3937. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Azminah; Mulia, K.; Yanuar, A.; Mun’im, A. Angiotensin-converting enzyme inhibitory activity of polyphenolic compounds from Peperomia pellucida (L) Kunth: An in silico molecular docking study. J. Appl. Pharm. Sci. 2019, 9, 25–31. [Google Scholar]
- Arya, H.; Syed, S.B.; Singh, S.S.; Ampasala, D.R.; Coumar, M.S. In Silico Investigations of Chemical Constituents of Clerodendrum colebrookianum in the Anti-Hypertensive Drug Targets: ROCK, ACE, and PDE5. Interdiscip. Sci. Comput. Life Sci. 2018, 10, 792–804. [Google Scholar] [CrossRef]
- Moorthy, N.S.H.N.; Brás, N.F.; Ramos, M.J.; Fernandes, P.A. Binding mode prediction and identification of new lead compounds from natural products as renin and angiotensin converting enzyme inhibitors. RSC Adv. 2014, 4, 19550–19568. [Google Scholar] [CrossRef]
- Abouelkheir, M.; El-Metwally, T.H. Dipeptidyl peptidase-4 inhibitors can inhibit angiotensin converting enzyme. Eur. J. Pharmacol. 2019, 862, 172638. [Google Scholar] [CrossRef] [PubMed]
- Saadaoui, I.; Krichen, F.; Ben Salah, B.; Ben Mansour, R.; Miled, N.; Bougatef, A.; Kossentini, M. Design, synthesis and biological evaluation of Schiff bases of 4-amino-1,2,4-triazole derivatives as potent angiotensin converting enzyme inhibitors and antioxidant activities. J. Mol. Struct. 2019, 1180, 344–354. [Google Scholar] [CrossRef]
- Ben Salah, B.; Hamzaoui, S.; Krichen, F.; Saadaoui, I.; Ben Mansour, R.; Miled, N.; Bougatef, A.; Kossentini, M. Design, synthesis of novel Triazolones and bis-Triazolones derivatives under ultrasound irradiation and evaluation as potent angiotensin converting enzyme (ACE) inhibitors. Bioorg. Chem. 2018, 76, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Manikandan, A.; Moharil, P.; Sathishkumar, M.; Muñoz-Garay, C.; Sivakumar, A. Therapeutic investigations of novel indoxyl-based indolines: A drug target validation and Structure-Activity Relationship of angiotensin-converting enzyme inhibitors with cardiovascular regulation and thrombolytic potential. Eur. J. Med. Chem. 2017, 141, 417–426. [Google Scholar] [CrossRef]
- Stoičkov, V.; Šarić, S.; Golubović, M.; Zlatanović, D.; Krtinić, D.; Dinić, L.; Mladenović, B.; Sokolović, D.; Veselinović, A.M. Development of non-peptide ACE inhibitors as novel and potent cardiovascular therapeutics: An in silico modelling approach. SAR QSAR Environ. Res. 2018, 29, 503–515. [Google Scholar] [CrossRef]
- Wallis, E.J.; Ramsay, L.E.; Hettiarachchi, J. Combined inhibition of neutral endopeptidase and angiotensin-converting enzyme by sampatrilat in essential hypertension. Clin. Pharmacol. Ther. 1998, 64, 439–449. [Google Scholar] [CrossRef]
- Sharma, R.K.; Espinoza-Moraga, M.; Poblete, H.; Douglas, R.G.; Sturrock, E.D.; Caballero, J.; Chibale, K. The Dynamic Nonprime Binding of Sampatrilat to the C-Domain of Angiotensin-Converting Enzyme. J. Chem. Inf. Model 2016, 56, 2486–2494. [Google Scholar] [CrossRef]
- Cozier, G.E.; Schwager, S.L.; Sharma, R.K.; Chibale, K.; Sturrock, E.D.; Acharya, K.R. Crystal structures of sampatrilat and sampatrilat-Asp in complex with human ACE—A molecular basis for domain selectivity. FEBS J. 2018, 285, 1477–1490. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, R.; Linusson, A.; Zamora, I. SHOP: Scaffold HOPping by GRID-based similarity searches. J. Med. Chem. 2007, 50, 2708–2717. [Google Scholar] [CrossRef]
- Bergmann, R.; Liljefors, T.; Sørensen, M.D.; Zamora, I. SHOP: Receptor-based scaffold HOPping by GRID-based similarity searches. J. Chem. Inf. Model 2009, 49, 658–669. [Google Scholar] [CrossRef]
- Douglas, R.G.; Sharma, R.K.; Masuyer, G.; Lubbe, L.; Zamora, I.; Acharya, K.R.; Chibale, K.; Sturrock, E.D. Fragment-based design for the development of N-domain-selective angiotensin-1-converting enzyme inhibitors. Clin. Sci. 2014, 126, 305–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fienberg, S.; Cozier, G.E.; Acharya, K.R.; Chibale, K.; Sturrock, E.D. The Design and Development of a Potent and Selective Novel Diprolyl Derivative That Binds to the N-Domain of Angiotensin-I Converting Enzyme. J. Med. Chem. 2018, 61, 344–359. [Google Scholar] [CrossRef] [PubMed]
- GOSTAR Drug Database and Clinical Candidate Database; Excelra Knowledge Solutions: Hyderabad, India. Available online: https://www.gostardb.com/index.jsp (accessed on 22 July 2013).
- Velázquez-Libera, J.L.; Navarro-Retamal, C.; Caballero, J. Insights into the Structural Requirements of 2(S)-Amino-6-Boronohexanoic Acid Derivatives as Arginase I Inhibitors: 3D-QSAR, Docking, and Interaction Fingerprint Studies. Int. J. Mol. Sci. 2018, 19, 2956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero, J.; Morales-Bayuelo, A.; Navarro-Retamal, C. Mycobacterium tuberculosis serine/threonine protein kinases: Structural information for the design of their specific ATP-competitive inhibitors. J. Comput. Aided Mol. Des. 2018, 32, 1315–1336. [Google Scholar] [CrossRef] [PubMed]
- Velázquez-Libera, J.L.; Rossino, G.; Navarro-Retamal, C.; Collina, S.; Caballero, J. Docking, Interaction Fingerprint, and Three-Dimensional Quantitative Structure-Activity Relationship (3D-QSAR) of Sigma1 Receptor Ligands, Analogs of the Neuroprotective Agent RC-33. Front Chem. 2019, 7, 496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velázquez-Libera, J.L.; Murillo-López, J.A.; de la Torre, A.F.; Caballero, J. Structural Requirements of N-alpha-Mercaptoacetyl Dipeptide (NAMdP) Inhibitors of Pseudomonas Aeruginosa Virulence Factor LasB: 3D-QSAR, Molecular Docking, and Interaction Fingerprint Studies. Int. J. Mol. Sci. 2019, 20, 6133. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.; Chuaqui, C.; Singh, J. Structural interaction fingerprint (SIFt): A novel method for analyzing three-dimensional protein-ligand binding interactions. J. Med. Chem. 2004, 47, 337–344. [Google Scholar] [CrossRef]
- Singh, J.; Deng, Z.; Narale, G.; Chuaqui, C. Structural interaction fingerprints: A new approach to organizing, mining, analyzing, and designing protein-small molecule complexes. Chem. Biol. Drug Des. 2006, 67, 5–12. [Google Scholar] [CrossRef]
- Watermeyer, J.M.; Kröger, W.L.; O’Neill, H.G.; Sewell, B.T.; Sturrock, E.D. Characterization of domain-selective inhibitor binding in angiotensin-converting enzyme using a novel derivative of lisinopril. Biochem. J. 2010, 428, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Nchinda, A.T.; Chibale, K.; Redelinghuys, P.; Sturrock, E.D. Synthesis and molecular modeling of a lisinopril-tryptophan analogue inhibitor of angiotensin I-converting enzyme. Bioorg. Med. Chem. Lett. 2006, 16, 4616–4619. [Google Scholar] [CrossRef]
- Natesh, R.; Schwager, S.L.U.; Sturrock, E.D.; Acharya, K.R. Crystal structure of the human angiotensin-converting enzyme–lisinopril complex. Nature 2003, 421, 551–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watermeyer, J.M.; Kröger, W.L.; O’Neill, H.G.; Sewell, B.T.; Sturrock, E.D. Probing the basis of domain-dependent inhibition using novel ketone inhibitors of Angiotensin-converting enzyme. Biochemistry 2008, 47, 5942–5950. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein–Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes, E.; Badimon, L.; Caballero, J.; Padró, T.; Vilahur, G.; Alarcón, M.; Pérez, P.; Palomo, I. Protective mechanisms of adenosine 5′-monophosphate in platelet activation and thrombus formation. Thromb. Haemost. 2014, 111, 491–507. [Google Scholar] [CrossRef]
- Fuentes, E.; Pereira, J.; Mezzano, D.; Alarcón, M.; Caballero, J.; Palomo, I. Inhibition of platelet activation and thrombus formation by adenosine and inosine: Studies on their relative contribution and molecular modeling. PLoS ONE 2014, 9, e112741. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, E.; Caballero, J.; Alarcón, M.; Rojas, A.; Palomo, I. Chlorogenic Acid Inhibits Human Platelet Activation and Thrombus Formation. PLoS ONE 2014, 9, e90699. [Google Scholar] [CrossRef] [Green Version]
- Quesada-Romero, L.; Mena-Ulecia, K.; Tiznado, W.; Caballero, J. Insights into the interactions between maleimide derivates and GSK3β combining molecular docking and QSAR. PLoS ONE 2014, 9, e102212. [Google Scholar] [CrossRef]
- Muñoz-Gutierrez, C.; Adasme-Carreño, F.; Fuentes, E.; Palomo, I.; Caballero, J. Computational study of the binding orientation and affinity of PPARγ agonists: Inclusion of ligand-induced fit by cross-docking. RSC Adv. 2016, 6, 64756–64768. [Google Scholar] [CrossRef]
- Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | PDB | d[C3] (Å) 1 | d[C2] (Å) 2 | d[VC2] (Å) 3 | d[VC1] (Å) 4 | d[C1] (Å) 5 |
|---|---|---|---|---|---|---|
| nACE | ||||||
| 1 | (6EN5//6F9V) | - | 3.1//3.2 | 1.9//2.0 | 2.9/2.9//2.7/2.9 | 2.7//3.0 |
| 2 | (6F9V) | 2.9 | - | 2.9 | 3.2/2.8 | 3.1 |
| 3 | (6EN5) | - | 3.1 | 1.9 | 2.9/2.8 | 2.9 |
| cACE | ||||||
| 1 | (6F9T//6F9U) | - | 3.0//3.2 | 1.9//2.1 | 7.6 6/3.2//4.1/2.7 | 2.8//2.8 |
| 2 | (6F9U) | 2.6 | - | 2.0 | 2.5/2.7 | 3.2 |
| 3 | (6F9T) | 3.1 | - | 1.9 | 8.6 6/3.2 | 2.8 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caballero, J. Considerations for Docking of Selective Angiotensin-Converting Enzyme Inhibitors. Molecules 2020, 25, 295. https://doi.org/10.3390/molecules25020295
Caballero J. Considerations for Docking of Selective Angiotensin-Converting Enzyme Inhibitors. Molecules. 2020; 25(2):295. https://doi.org/10.3390/molecules25020295
Chicago/Turabian StyleCaballero, Julio. 2020. "Considerations for Docking of Selective Angiotensin-Converting Enzyme Inhibitors" Molecules 25, no. 2: 295. https://doi.org/10.3390/molecules25020295
APA StyleCaballero, J. (2020). Considerations for Docking of Selective Angiotensin-Converting Enzyme Inhibitors. Molecules, 25(2), 295. https://doi.org/10.3390/molecules25020295