Tautomerism and Self-Association in the Solution of New Pinene-Bipyridine and Pinene-Phenanthroline Derivatives

 , , ,
, , , 
 , and
, and
Abstract
:1. Introduction
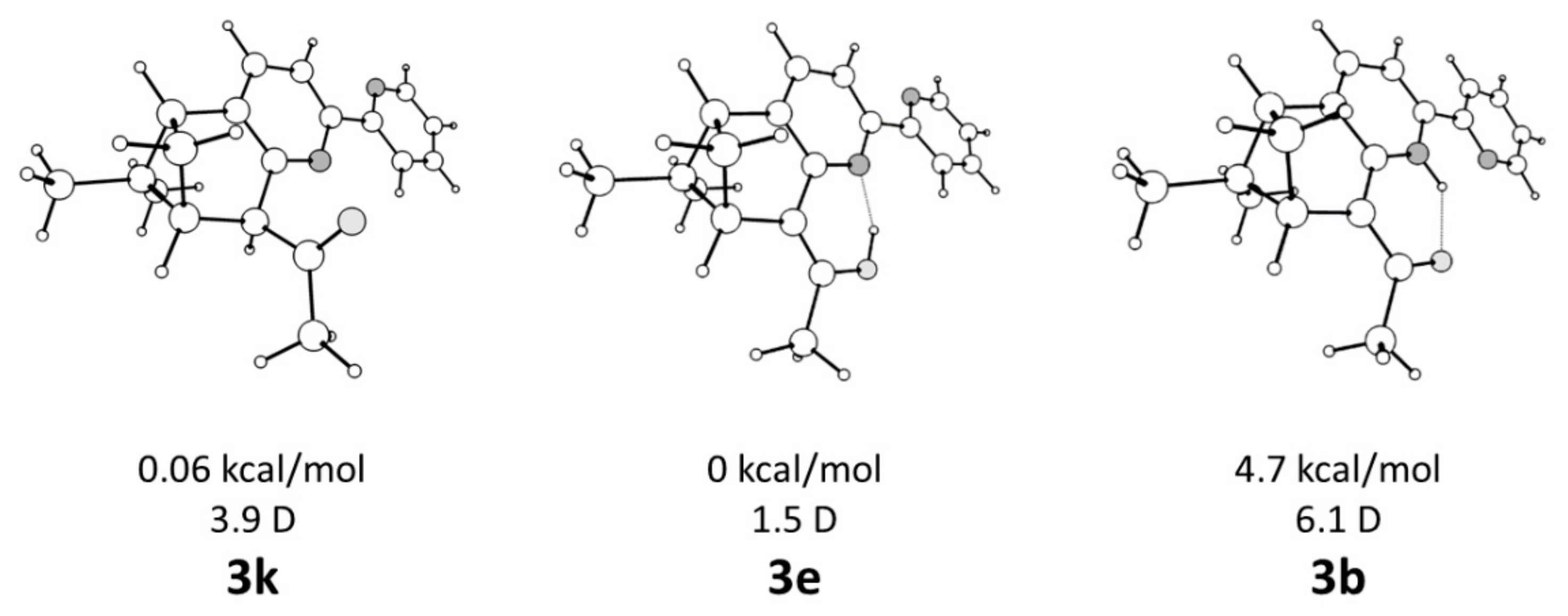
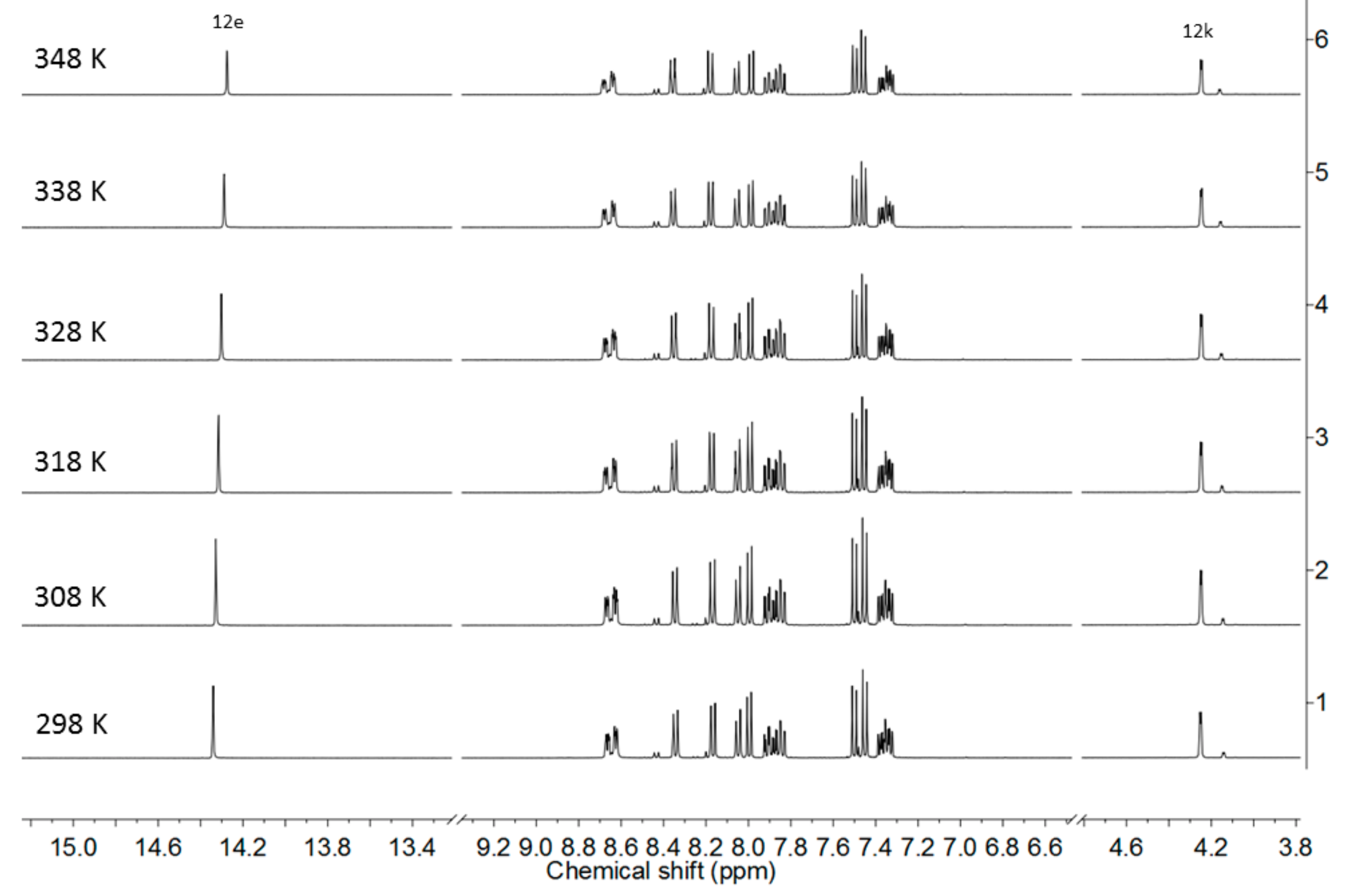
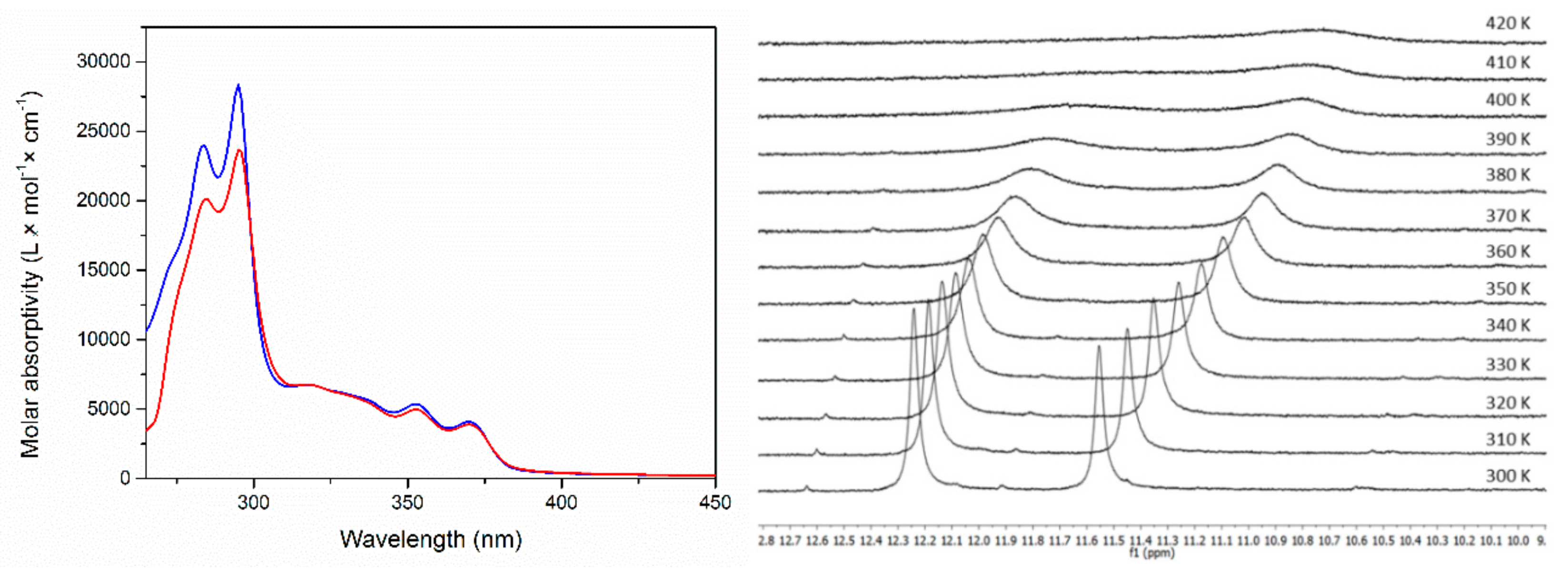
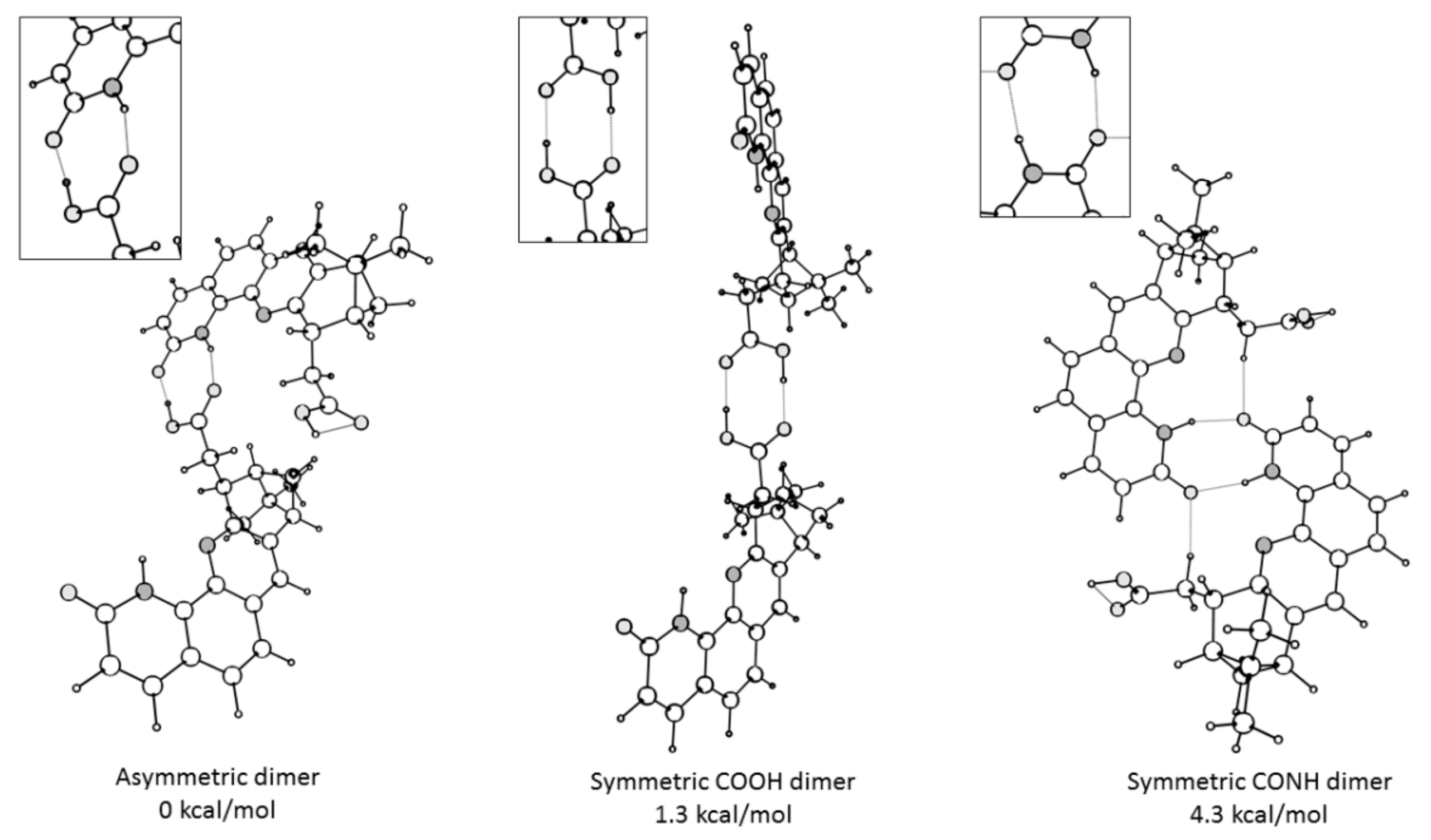
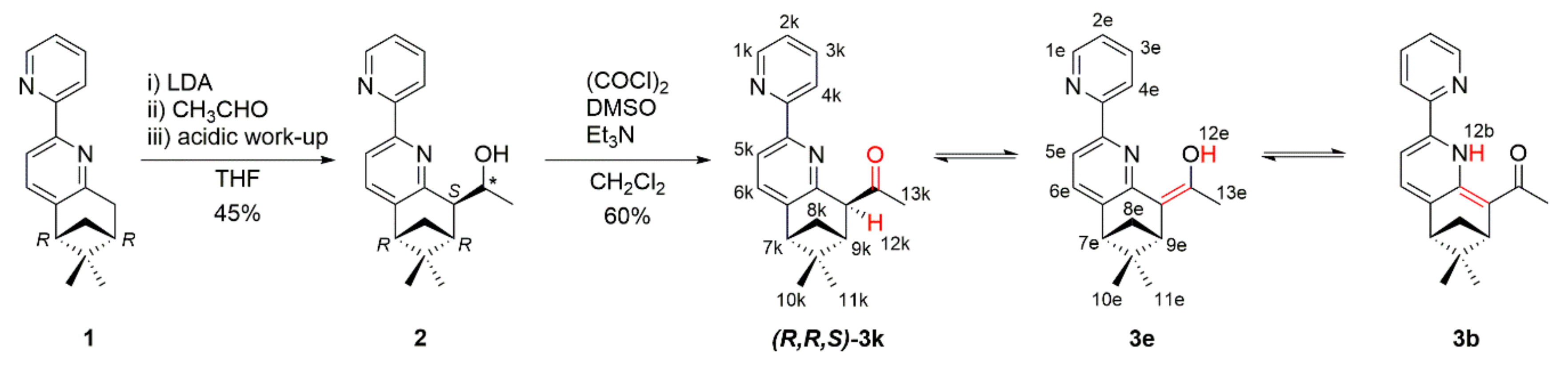
2. Results and Discussion
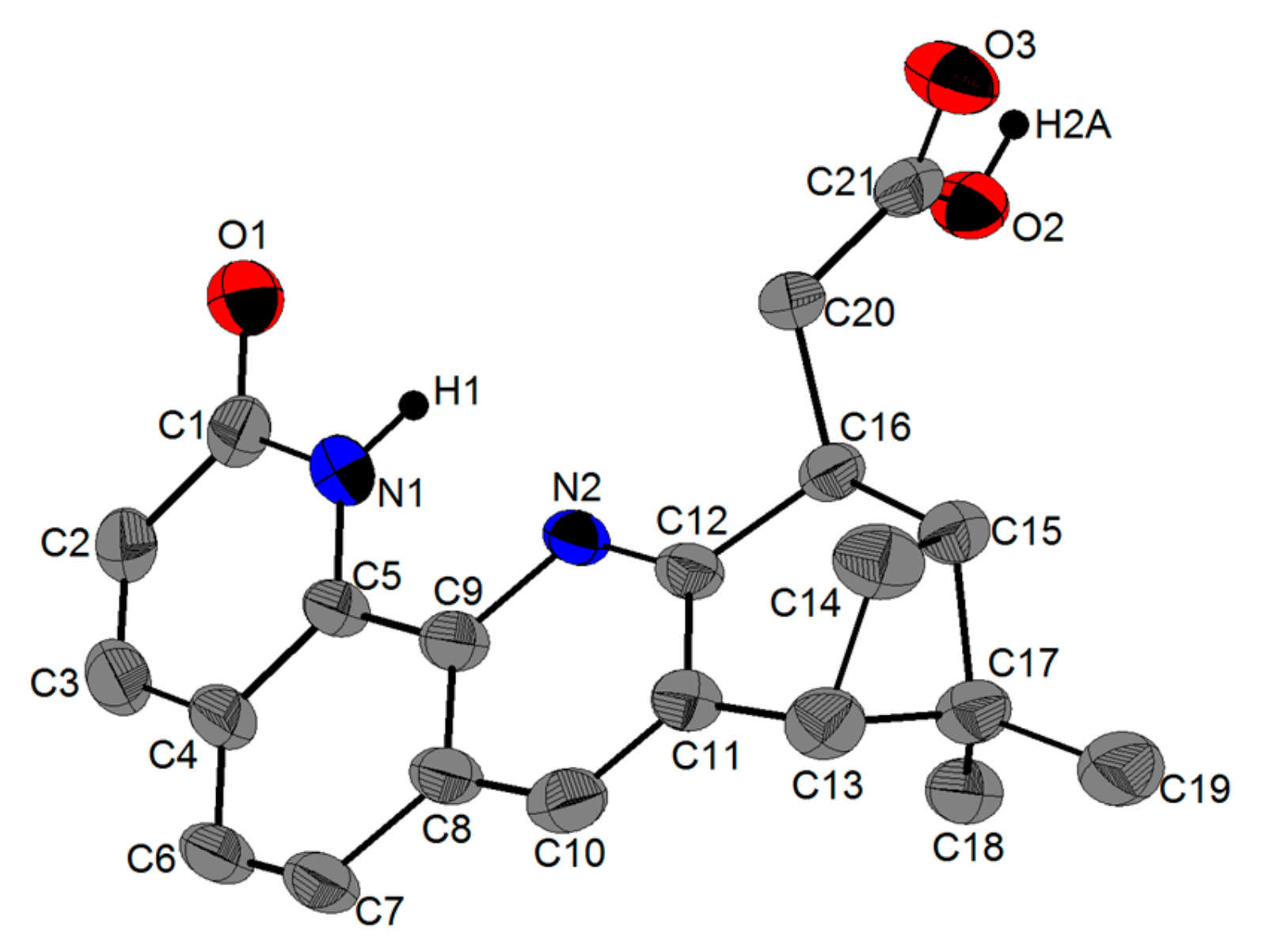
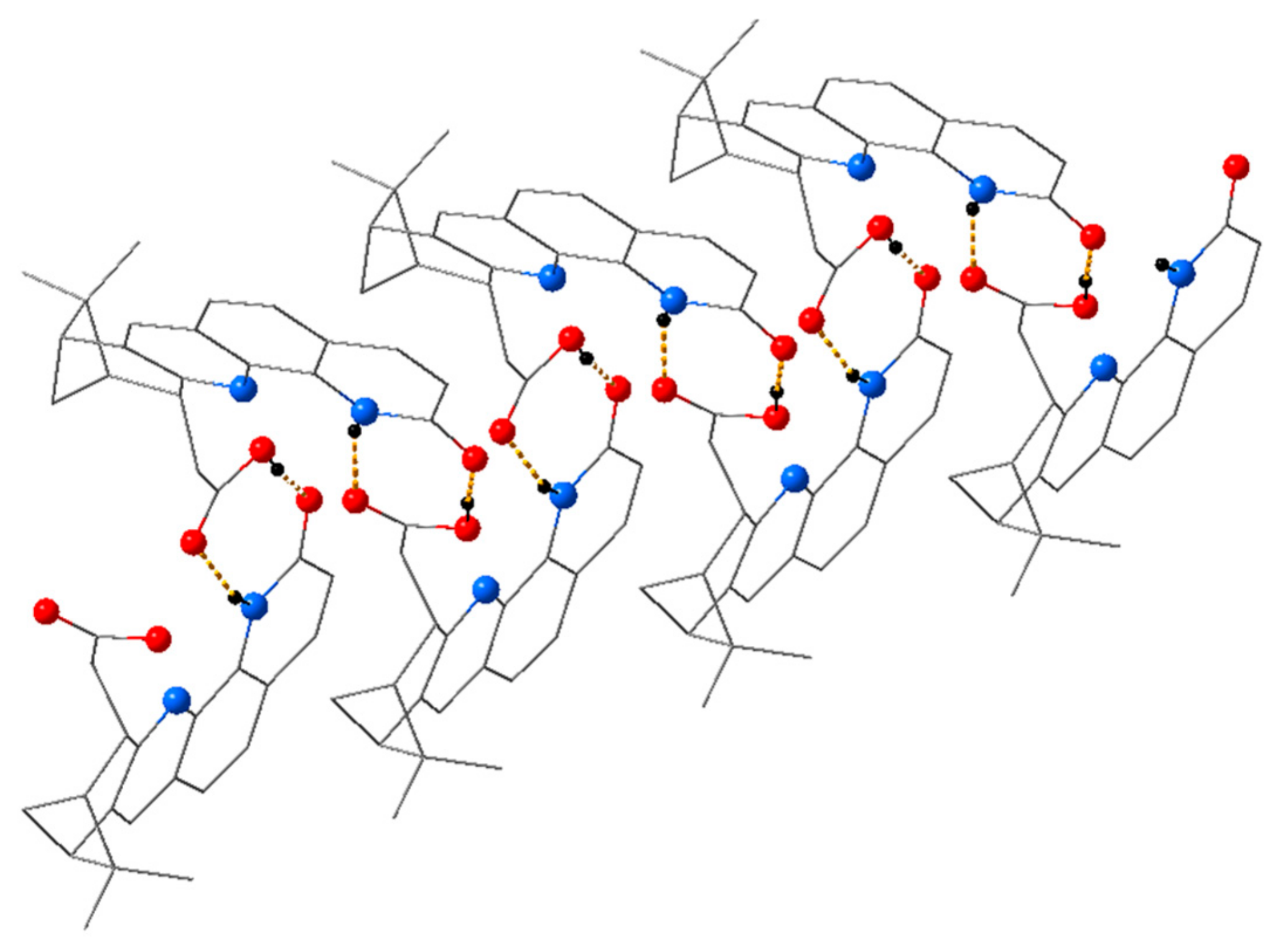
Single Crystal X-Ray Diffraction Studies
3. Materials and Methods
3.1. General
3.1.1. Single Crystal X-Ray Diffraction of 6
3.1.2. Synthesis of 2
3.1.3. Synthesis of 3
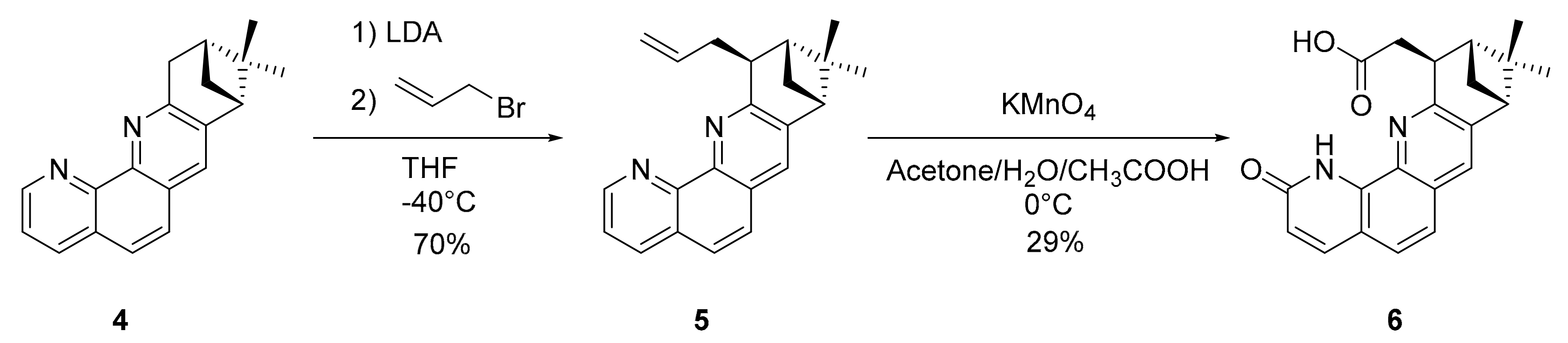
3.1.4. Synthesis of 5
3.1.5. Synthesis of 6
3.1.6. Quantum-Chemical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lama, M.; Mamula, O.; Kottas, G.S.; De Cola, L.; Stoeckli-Evans, H.; Shova, S. Enantiopure, Supramolecular Helices Containing Three-Dimensional Tetranuclear Lanthanide (III) Arrays: Synthesis, Structure, Properties, and Solvent-Driven Trinuclear/Tetranuclear Interconversion. Inorg. Chem. 2008, 47, 8000–8015. [Google Scholar] [CrossRef] [PubMed]
- Von Zelewsky, A.; Mamula, O. The bright future of stereoselective synthesis of co-ordination compounds. J. Chem. Soc. Dalton Trans. 2000, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Mamula, O.; Von Zelewsky, A.; Bark, T.; Bernardinelli, G. Stereoselective Synthesis of Coordination Compounds: Self-Assembly of a Polymeric Double Helix with Controlled Chirality. Angew. Chem. Int. Ed. 1999, 38, 2945–2948. [Google Scholar] [CrossRef]
- Perret-Aebi, L.-E.; von Zelewsky, A.; Neels, A. Diastereoselective preparation of Cu(I) and Ag(I) double helices by the use of chiral bis-bipyridine ligands. New J. Chem. 2009, 33, 462–465. [Google Scholar] [CrossRef] [Green Version]
- Mamula, O.; Bark, T.; Quinodoz, B.; Stoeckli-Evans, H.; von Zelewsky, A. Self-assembly of Ag(I) helicates with new enantiopure 5,6-Chiragen type ligands. Inorg. Chim. Acta 2018, 475, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Rich, J.; Rodríguez, M.; Romero, I.; Vaquer, L.; Sala, X.; Llobet, A.; Corbella, M.; Collomb, M.-N.; Fontrodona, X. Mn(ii) complexes containing the polypyridylic chiral ligand (−)-pinene [5,6]bipyridine. Catalysts for oxidation reactions. Dalton Trans. 2009, 38, 8117–8126. [Google Scholar] [CrossRef]
- Chelucci, G.; Saba, A. New 1,10-phenanthroline ligands for asymmetric catalysis: Enantioselective palladium catalyzed allylic substitution. Tetrahedron Asymmetry 1998, 9, 2575–2578. [Google Scholar] [CrossRef]
- Schmitz, C.; Leitner, W.; Franciò, G. Pinene-derived monodentate phosphoramidites for asymmetric hydrogenation. Eur. J. Org. Chem. 2015, 2015, 2889–2901. [Google Scholar] [CrossRef]
- Solea, A.B.; Wohlhauser, T.; Abbasi, P.; Mongbanziama, Y.; Crochet, A.; Fromm, K.M.; Novitchi, G.; Train, C.; Pilkington, M.; Mamula, O. Versatile synthesis of chiral 6-oxoverdazyl radical ligands-new building blocks for multifunctional molecule-based magnets. Dalton Trans. 2018, 47, 4785–4789. [Google Scholar] [CrossRef] [Green Version]
- Hayoz, P.; Von Zelewsky, A. New versatile optically active bipyridines as building blocks for helicating and caging ligands. Tetrahedron Lett. 1992, 33, 5165–5168. [Google Scholar] [CrossRef]
- Lama, M.; Mamula, O.; Scopelliti, R. Enantiopure Oxygen-Functionalized Bipyridine Derivatives: Synthesis and Characterization. Synlett 2004, 10, 1808–1810. [Google Scholar]
- Malkov, A.V.; Baxendale, I.R.; Bella, M.; Langer, V.; Fawcett, J.; Russell, D.R.; Mansfield, D.J.; Valko, M.; Kočovský, P. Synthesis of New Chiral 2,2′-Bipyridyl-Type Ligands, Their Coordination to Molybdenum(0), Copper(II), and Palladium(II), and Application in Asymmetric Allylic Substitution, Allylic Oxidation, and Cyclopropanation. Organometallics 2001, 20, 673–690. [Google Scholar] [CrossRef]
- Chelucci, G.; Saba, A.; Sanna, G.; Soccolini, F. Chiral 2,2′-bipyridines, 5,6-dihydro-1,10-phenanthrolines and 1,10-phenanthrolines as ligands for enantioselective palladium catalyzed allylic substitution. Tetrahedron Asymmetry 2000, 11, 3427–3438. [Google Scholar] [CrossRef]
- Kumbhar, S.V.; Chen, C. Enantioselective Nozaki-Hiyama-Kishi allylation-lactonization for the syntheses of 3-substituted phthalides. Rsc Adv. 2018, 8, 41355–41357. [Google Scholar] [CrossRef] [Green Version]
- Hayoz, P.; von Zelewsky, A.; Stoeckli-Evans, H. Stereoselective Synthesis of Octahedral Complexes with Predetermined Helical Chirality. J. Am. Chem. Soc. 1993, 115, 5111–5114. [Google Scholar] [CrossRef]
- Mürner, H.; Von Zelewsky, A.; Stoeckli-Evans, H. Octahedral complexes with predetermined helical chirality: Xylene-bridged bis([4,5]-pineno-2,2′-bipyridine) ligands (chiragen[o-, m-, p-xyl]) with ruthenium(II). Inorg. Chem. 1996, 35, 3931–3935. [Google Scholar] [CrossRef]
- Murner, H.; von Zelewsky, A.; Hopfgartner, G. Dinuclear metal complexes of Cd(II), Zn(II) and Fe(II) with triple-helical structure and predetermined chirality. Inorg. Chim. Acta 1998, 271, 36–39. [Google Scholar] [CrossRef]
- Mamula, O.; Monlien, F.J.; Porquet, A.; Hopfgartner, G.; Merbach, A.E.; Von Zelewsky, A. Self-assembly of multinuclear coordination species with chiral bipyridine ligands: Silver complexes of 5,6-CHIRAGEN (o,m,p-xylidene) ligands and equilibrium behaviour in solution. Chem. Eur. J. 2001, 7, 533–539. [Google Scholar] [CrossRef]
- Mürner, H.; Belser, P.; Von Zelewsky, A. New configurationally stable chiral building blocks for polynuclear coordination compounds: Ru(chiragen[X])Cl2. J. Am. Chem. Soc. 1996, 118, 7989–7994. [Google Scholar] [CrossRef]
- Gladiali, S.; Chelucci, G.; Mudadu, M.S.; Gastaut, M.A.; Thummel, R.P. Friedländer synthesis of chiral alkyl-substituted 1,10-phenanthrolines. J. Org. Chem. 2001, 66, 400–405. [Google Scholar] [CrossRef]
- Chelucci, G.; Loriga, G.; Murineddu, G.; Pinna, G.A. Synthesis of chiral C2-symmetric 1,10-phenanthrolines from naturally occurring monoterpenes. Synthesis 2003, 73–78. [Google Scholar] [CrossRef]
- Gawinecki, R.; Kolehmainen, E.; Loghmani-Khouzani, H.; Ośmiałowski, B.; Lovász, T.; Rosa, P. Effect of π-electron delocalization on tautomeric equilibria - Benzoannulated 2-phenacylpyridines. Eur. J. Org. Chem. 2006, 2817–2824. [Google Scholar] [CrossRef]
- Antonov, L.; Fabian, W.M.F.; Taylor, P.J. Tautomerism in some aromatic Schiff bases and related azo compounds: An LSER study. J. Phys. Org. Chem. 2005, 18, 1169–1175. [Google Scholar] [CrossRef]
- Skotnicka, A.; Czeleń, P.; Gawinecki, R. Tautomeric Equilibria in Solutions of 2-Phenacylbenzimidazoles. Heteroat. Chem. 2019. [Google Scholar] [CrossRef] [Green Version]
- Pol, P.D.; Mahesh, R.T.; Nandibewoor, S.T. Kinetics and mechanism of chromium(III) catalyzed oxidation of 1,10-phenanthroline by alkaline permanganate. React. Kinet. Catal. Lett. 2004, 81, 113–119. [Google Scholar] [CrossRef]
- Mason, S.F. The tautomerism of N-heteroaromatic hydroxy-compounds. Part I. Infrared spectra. J. Chem. Soc. 1957, 980, 4874–4880. [Google Scholar] [CrossRef]
- Dey, A.S.; Joullié, M.M. Synthesis and properties of fluorine-containing heterocyclic compounds. II. trifluoromethyl benzo[h]quinolines, benzo[h]-1,6-napthyridines, 1,7- and 1,10-phenanthrolines. J. Heterocycl. Chem. 1965, 2, 120–125. [Google Scholar] [CrossRef]
- Goldsmith, M.R.; Jayasuriya, N.; Beratan, D.N.; Wipf, P. Optical Rotation of Noncovalent Aggregates. J. Am. Chem. Soc. 2003, 125, 15696–15697. [Google Scholar] [CrossRef]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002, 41, 49–76. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. Sect. A Found. Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Bulik, I.W.; Scalmani, G.; Frisch, M.J.; Scuseria, G.E. Noncollinear density functional theory having proper invariance and local torque properties. Phys. Rev. B 2013, 87, 035117. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Chem. Acc. 2007, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Kawauchi, S.; Antonov, L. Description of the Tautomerism in Some Azonaphthols. J. Phys. Org. Chem. 2013, 26, 643–652. [Google Scholar] [CrossRef]
- Manolova, Y.; Kurteva, V.; Antonov, L.; Marciniak, H.; Lochbrunner, S.; Crochet, A.; Fromm, K.M.; Kamounah, F.S.; Hansen, P.E. 4-Hydroxy-1-naphthaldehydes: Proton transfer or deprotonation. Phys. Chem. Chem. Phys. 2015, 17, 10238–10249. [Google Scholar] [CrossRef] [Green Version]
- Manolova, Y.; Marciniak, H.; Tschierlei, S.; Fennel, F.; Kamounah, F.S.; Lochbrunner, S.; Antonov, L. Solvent control of intramolecular proton transfer: Is 4-hydroxy-3-(piperidin-1-ylmethyl)-1-naphthaldehyde a proton crane? Phys. Chem. Chem. Phys. 2017, 19, 7316–7325. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Improta, R. UV-Visible Absorption and Emission Energies in Condensed Phase by PCM/TD-DFT Methods. Comput. Strat. Spectrosc. 2011, 37–75. [Google Scholar]
- Kawauchi, S.; Antonov, L.; Okuno, Y. Prediction of the color of dyes by using time-dependent density functional theory (TD-DFT). Bulg. Chem. Commun. 2014, 46, 228–237. [Google Scholar]
- Jacquemin, D.; Mennucci, B.; Adamo, C. Excited-state calculations with TD-DFT: From benchmarks to simulations in complex environments. Phys. Chem. Chem. Phys. 2011, 13, 16987. [Google Scholar] [CrossRef] [PubMed]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Deneva, V.; Lyčka, A.; Hristova, S.; Crochet, A.; Fromm, K.M.; Antonov, L. Tautomerism in azo dyes: Border cases of azo and hydrazo tautomers as possible NMR reference compounds. Dye. Pigm. 2019, 165, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Simon, S.; Duran, M.; Dannenberg, J.J. How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? J. Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds 1–6 are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Theoretical ΔE (kcal/mol) | Experimental ΔG° (kcal/mol) |
|---|---|---|
| CD3CN | 0.84 | 0.15 |
| CD2Cl2 | 0.37 | 0.28 |
| CDCl3 | −0.06 | 0.19 |
| decalin-d18 | −0.82 | −0.46 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solea, A.B.; Cornu, I.; Deneva, V.; Crochet, A.; Fromm, K.M.; Antonov, L.; Allemann, C.; Mamula, O. Tautomerism and Self-Association in the Solution of New Pinene-Bipyridine and Pinene-Phenanthroline Derivatives. Molecules 2020, 25, 298. https://doi.org/10.3390/molecules25020298
Solea AB, Cornu I, Deneva V, Crochet A, Fromm KM, Antonov L, Allemann C, Mamula O. Tautomerism and Self-Association in the Solution of New Pinene-Bipyridine and Pinene-Phenanthroline Derivatives. Molecules. 2020; 25(2):298. https://doi.org/10.3390/molecules25020298
Chicago/Turabian StyleSolea, Atena B., Ivan Cornu, Vera Deneva, Aurelien Crochet, Katharina M. Fromm, Liudmil Antonov, Christophe Allemann, and Olimpia Mamula. 2020. "Tautomerism and Self-Association in the Solution of New Pinene-Bipyridine and Pinene-Phenanthroline Derivatives" Molecules 25, no. 2: 298. https://doi.org/10.3390/molecules25020298
APA StyleSolea, A. B., Cornu, I., Deneva, V., Crochet, A., Fromm, K. M., Antonov, L., Allemann, C., & Mamula, O. (2020). Tautomerism and Self-Association in the Solution of New Pinene-Bipyridine and Pinene-Phenanthroline Derivatives. Molecules, 25(2), 298. https://doi.org/10.3390/molecules25020298