Tautomerism in Azo and Azomethyne Dyes: When and If Theory Meets Experiment


Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Taylor, P.J.; van der Zwan, G.; Antonov, L. Tautomerism: Introduction, history, and recent developments in experimental and theoretical methods. In Tautomerism: Methods and Theories; Antonov, L., Ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 1–24. ISBN 978-3-527-65882-4. [Google Scholar]
- Baker, J.W. Tautomerism; George Routledge and Sons Ltd.: London, UK, 1934. [Google Scholar]
- Eistert, B. Tautomerie und Mesomerie: Gleichgewicht und ‘“Resonanz”; Enke: Stuttgart, Germany, 1938. [Google Scholar]
- Elguero, J.; Marzin, C.; Katritzky, A.R.; Linda, P. The tautomerism of heterocycles. In Advances in Heterocyclic Chemistry; Academic Press: New York, NY, USA, 1976; ISBN 978-0-12-020651-3. [Google Scholar]
- Minkin, V.I.; Olekhnovich, L.P.; Zhdanov, I.A. Molecular Design of Tautomeric Compounds; Springer: Dordrecht, The Netherlands, 1988; ISBN 978-94-009-1429-2. [Google Scholar]
- Elguero, J.; Katritzky, A.R.; Denisko, O.V. Prototropic tautomerism of heterocycles: Heteroaromatic tautomerism—General overview and methodology. In Advances in Heterocyclic Chemistry; Elsevier: Amesterdam, The Netherlands, 2000; Volume 76, pp. 1–84. ISBN 978-0-12-020776-3. [Google Scholar]
- Minkin, V.I.; Garnovskii, A.D.; Elguero, J.; Katritzky, A.R.; Denisko, O.V. The tautomerism of heterocycles: Five-membered rings with two or more heteroatoms. In Advances in Heterocyclic Chemistry; Elsevier: Amesterdam, The Netherlands, 2000; Volume 76, pp. 157–323. ISBN 978-0-12-020776-3. [Google Scholar]
- Hydrogen-Transfer Reactions, 1st ed.; Hynes, J.T.; Klinman, J.P.; Limbach, H.; Schowen, R.L. (Eds.) Wiley: Hoboken, NJ, USA, 2006; ISBN 978-3-527-30777-7. [Google Scholar]
- Stanovnik, B.; Tišler, M.; Katritzky, A.R.; Denisko, O.V. The tautomerism of heterocycles: Substituent tautomerism of six-membered ring heterocycles. In Advances in Heterocyclic Chemistry; Elsevier: Amesterdam, The Netherlands, 2006; Volume 91, pp. 1–134. ISBN 978-0-12-020791-6. [Google Scholar]
- Martin, Y.C. Let’s not forget tautomers. J. Comput.-Aided Mol. Des. 2009, 23, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Tautomerism: Methods and Theories; Antonov, L. (Ed.) Wiley-VCH: Weinheim, Germany, 2014; ISBN 978-3-527-33294-6. [Google Scholar]
- Tautomerism: Concepts and Applications in Science and Technology; Antonov, L. (Ed.) Wiley-VCH: Weinheim, Germany, 2016; ISBN 978-3-527-33995-2. [Google Scholar]
- Gordon, P.F.; Gregory, P. Azo dyes. In Organic Chemistry in Colour; Springer-Verlag: Berlin, Germany, 1987; pp. 95–162. ISBN 978-3-642-82959-8. [Google Scholar]
- Kelemen, J. Azo-hydrazone tautomerism in azo dyes. I. A comparative study of 1-phenylazo-2-naphthol and 1-phenylazo-2-naphthylamine derivatives by electronic spectroscopy. Dye. Pigment. 1981, 2, 73–91. [Google Scholar] [CrossRef]
- Zollinger, H. Color Chemistry: Syntheses, Properties, and Applications of Organic Dyes and Pigments, 3rd ed.; Wiley-VCH: Zürich, Switzerland; Weinheim, Germany, 2003; ISBN 978-3-906390-23-9. [Google Scholar]
- Christie, R.M. Colour Chemistry, 2nd ed.; Royal Society of Chemistry: Cambridge, UK, 2015; ISBN 978-1-84973-328-1. [Google Scholar]
- Zincke, T.; Bindewald, H. Ueber phenylhydrazinderivate des α- und β-naphtochinons. Identität des α-derivats mit dem azoderivat des α-naphtols. Ber. Der Dtsch. Chem. Ges. 1884, 17, 3026–3033. [Google Scholar] [CrossRef]
- Antonov, L.; Kurteva, V.; Crochet, A.; Mirolo, L.; Fromm, K.M.; Angelova, S. Tautomerism in 1-phenylazo-4-naphthols: Experimental results vs quantum-chemical predictions. Dye. Pigment. 2012, 92, 714–723. [Google Scholar] [CrossRef] [Green Version]
- Antonov, L.; Hansen, P.E.; van der Zwan, G. Comment on “Spectroscopic studies of keto-enol tautomeric equilibrium of azo dyes” by M. A. Rauf, S. Hisaindee and N. Saleh, RSC Adv. 2015, 5, 18097. RSC Adv. 2015, 5, 67165–67167. [Google Scholar] [CrossRef]
- Hadjoudis, E.; Mavridis, I.M. Photochromism and thermochromism of Schiff bases in the solid state: Structural aspects. Chem. Soc. Rev. 2004. [Google Scholar] [CrossRef] [PubMed]
- Raczyńska, E.D.; Kosińska, W.; Ośmiałowski, B.; Gawinecki, R. Tautomeric equilibria in relation to pi-electron delocalization. Chem. Rev. 2005, 105, 3561–3612. [Google Scholar] [CrossRef] [PubMed]
- Kajal, A.; Bala, S.; Kamboj, S.; Sharma, N.; Saini, V. Schiff bases: A versatile pharmacophore. J. Catal. 2013, 2013, 1–14. [Google Scholar] [CrossRef]
- Hansen, P.E. Methods to distinguish tautomeric cases from static ones. In Tautomerism Concepts and Applications in Science and Technology; Antonov, L., Ed.; Wiley-VCH: Weinheim, Germany, 2016; pp. 35–74. ISBN 978-3-527-69571-3. [Google Scholar]
- Fabian, W.M.F. Quantum chemical calculation of tautomeric equilibria. In Tautomerism; Antonov, L., Ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 337–368. ISBN 978-3-527-65882-4. [Google Scholar]
- Antonov, L.; Nedeltcheva, D. Resolution of overlapping UV–Vis absorption bands and quantitative analysis. Chem. Soc. Rev. 2000, 29, 217–227. [Google Scholar] [CrossRef]
- Nedeltcheva, D.; Antonov, L.; Lycka, A.; Damyanova, B.; Popov, S. Chemometric models for quantitative analysis of tautomeric Schiff bases and azo dyes. Curr. Org. Chem. 2009, 13, 217–240. [Google Scholar] [CrossRef]
- Kawauchi, S.; Antonov, L. Description of the tautomerism in some azonaphthols. J. Phys. Org. Chem. 2013, 26, 643–652. [Google Scholar] [CrossRef]
- Neese, F.; Hansen, A.; Wennmohs, F.; Grimme, S. Accurate theoretical chemistry with coupled pair models. Acc. Chem. Res. 2009, 42, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Waller, M.P. JACOB: A dynamic database for computational chemistry benchmarking. J. Chem. Inf. Model. 2012, 52, 3255–3262. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Truhlar, D.G. Quest for a universal density functional: The accuracy of density functionals across a broad spectrum of databases in chemistry and physics. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2014, 372, 20120476. [Google Scholar] [CrossRef] [PubMed]
- Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, N.; Casanova-Páez, M.; Goerigk, L. Semi-empirical or non-empirical double-hybrid density functionals: Which are more robust? Phys. Chem. Chem. Phys. 2018, 20, 23175–23194. [Google Scholar] [CrossRef] [PubMed]
- Morgante, P.; Peverati, R. ACCDB: A Collection of chemistry databases for broad computational purposes. J. Comput. Chem. 2019, 40, 839–848. [Google Scholar] [CrossRef]
- Okawara, M.; Kitao, T.; Hirashima, T.; Matsuoka, M. Organic Colorants a Handbook of Data of Selected Dyes for Electro-Optical Applications; Kodansha: Tokyo, Japan; Elsevier: Amsterdam, The Netherlands, 1988; ISBN 978-0-444-98884-3. [Google Scholar]
- Savarese, M.; Brémond, É.; Antonov, L.; Ciofini, I.; Adamo, C. Computational insights into excited-state proton-transfer reactions in azo and azomethine dyes. ChemPhysChem 2015, 16, 3966–3973. [Google Scholar] [CrossRef]
- Antonov, L.; Fabian, W.M.F.; Taylor, P.J. Tautomerism in some aromatic Schiff bases and related azo compounds: An LSER study. J. Phys. Org. Chem. 2005, 18, 1169–1175. [Google Scholar] [CrossRef]
- Nedeltcheva, D.; Antonov, L. Relative strength of the intramolecular hydrogen bonding in 1-phenylazo-naphthalen-2-ol and 1-phenyliminomethyl-naphtahlen-2-ol. J. Phys. Org. Chem. 2009, 22, 274–281. [Google Scholar] [CrossRef]
- Stoyanov, S.; Antonov, L. Quantitative analysis of azo-quinonehydrazone tautomeric equilibrium. Dye. Pigment. 1989, 10, 33–45. [Google Scholar] [CrossRef]
- Nedeltcheva, D.; Kurteva, V.; Topalova, I. Gas-phase tautomerism in hydroxy azo dyes—From 4-phenylazo-1-phenol to 4-phenylazo-anthracen-1-ol: Gas-phase tautomerism in hydroxy azo dyes. Rapid Commun. Mass Spectrom. 2010, 24, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, J. Developments in the Chemistry and Technology of Organic Dyes; Blackwell Scientific Publications: Oxford, UK; Boston, MA, USA, 1984; ISBN 978-0-632-01304-3. [Google Scholar]
- Fabian, W.M.F.; Antonov, L.; Nedeltcheva, D.; Kamounah, F.S.; Taylor, P.J. Tautomerism in hydroxynaphthaldehyde anils and azo analogues: A combined experimental and computational study. J. Phys. Chem. A 2004, 108, 7603–7612. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. Screened-exchange density functionals with broad accuracy for chemistry and solid-state physics. Phys. Chem. Chem. Phys. 2012, 14, 16187. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Roothaan, C.C.J. New developments in molecular orbital theory. Rev. Mod. Phys. 1951, 23, 69–89. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. Communication: A global hybrid generalized gradient approximation to the exchange-correlation functional that satisfies the second-order density-gradient constraint and has broad applicability in chemistry. J. Chem. Phys. 2011, 135, 191102. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. Improving the accuracy of hybrid meta-gga density functionals by range separation. J. Phys. Chem. Lett. 2011, 2, 2810–2817. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Henderson, T.M.; Izmaylov, A.F.; Scuseria, G.E.; Savin, A. Assessment of a middle-range hybrid functional. J. Chem. Theory Comput. 2008, 4, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Perdew, J.P. Generalized gradient approximation to the angle- and system-averaged exchange hole. J. Chem. Phys. 1998, 109, 3313–3320. [Google Scholar] [CrossRef]
- Austin, A.; Petersson, G.A.; Frisch, M.J.; Dobek, F.J.; Scalmani, G.; Throssell, K. A density functional with spherical atom dispersion terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom—Atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef] [PubMed]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef]
- Vydrov, O.A.; Scuseria, G.E. Assessment of a long-range corrected hybrid functional. J. Chem. Phys. 2006, 125, 234109. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Xu, X.; Goddard, W.A. From the cover: The X3LYP extended density functional for accurate descriptions of nonbond interactions, spin states, and thermochemical properties. Proc. Natl. Acad. Sci. USA 2004, 101, 2673–2677. [Google Scholar] [CrossRef] [PubMed]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Becke, A.D. Density functional calculations of molecular bond energies. J. Chem. Phys. 1986, 84, 4524–4529. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129–12137. [Google Scholar] [CrossRef]
- Boese, A.D.; Handy, N.C. New exchange-correlation density functionals: The role of the kinetic-energy density. J. Chem. Phys. 2002, 116, 9559–9569. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta–generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91. [Google Scholar] [CrossRef]
- Handy, N.C.; Cohen, A.J. Left-right correlation energy. Mol. Phys. 2001, 99, 403–412. [Google Scholar] [CrossRef]
- Cohen, A.J.; Handy, N.C. Dynamic correlation. Mol. Phys. 2001, 99, 607–615. [Google Scholar] [CrossRef]
- Boese, A.D.; Martin, J.M.L. Development of density functionals for thermochemical kinetics. J. Chem. Phys. 2004, 121, 3405–3416. [Google Scholar] [CrossRef] [Green Version]
- Crochet, A.; Fromm, K.M.; Kurteva, V.; Antonov, L. (E)-1-(4-Methoxyanthracen-1-yl)-2-phenyldiazene. Acta Crystallogr. Sect. E Struct. Rep. Online 2011, 67, o993. [Google Scholar] [CrossRef] [PubMed]
- Gilli, P.; Bertolasi, V.; Pretto, L.; Antonov, L.; Gilli, G. Variable-temperature X-ray crystallographic and DFT computational study of the N-H···O/N···H-O tautomeric competition in 1-(Arylazo)-2-naphthols. Outline of a transiton-state hydrogen-bond theory. J. Am. Chem. Soc. 2005, 127, 4943–4953. [Google Scholar] [CrossRef] [PubMed]
- Nedeltcheva, D.; Kamounah, F.S.; Mirolo, L.; Fromm, K.M.; Antonov, L. Solid state tautomerism in 2-((phenylimino)methyl)naphthalene-1-ol. Dye. Pigment. 2009, 83, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Antonov, L. Absorption UV-Vis Spectroscopy and chemometrics: from qualitative conclusions to quantitative analysis. In Tautomerism: Methods and Theories; Antonov, L., Ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 25–47. ISBN 978-3-527-65882-4. [Google Scholar]
- Antonov, L.; Fabian, W.M.F.; Nedeltcheva, D.; Kamounah, F.S. Tautomerism of 2-hydroxynaphthaldehyde Schiff bases. J. Chem. Soc. Perkin Trans. 2 2000, 1173–1179. [Google Scholar] [CrossRef]
- Ali, S.T.; Antonov, L.; Fabian, W.M.F. Phenol–quinone tautomerism in (Arylazo)naphthols and the analogous Schiff bases: Benchmark calculations. J. Phys. Chem. A 2014, 118, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Scuseria, G.E.; Schaefer, H.F. Is coupled cluster singles and doubles (CCSD) more computationally intensive than quadratic configuration interaction (QCISD)? J. Chem. Phys. 1989, 90, 3700–3703. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Nelder, J.A.; Mead, R. A Simplex method for function minimization. Comput. J. 1965, 7, 308–313. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the author. |




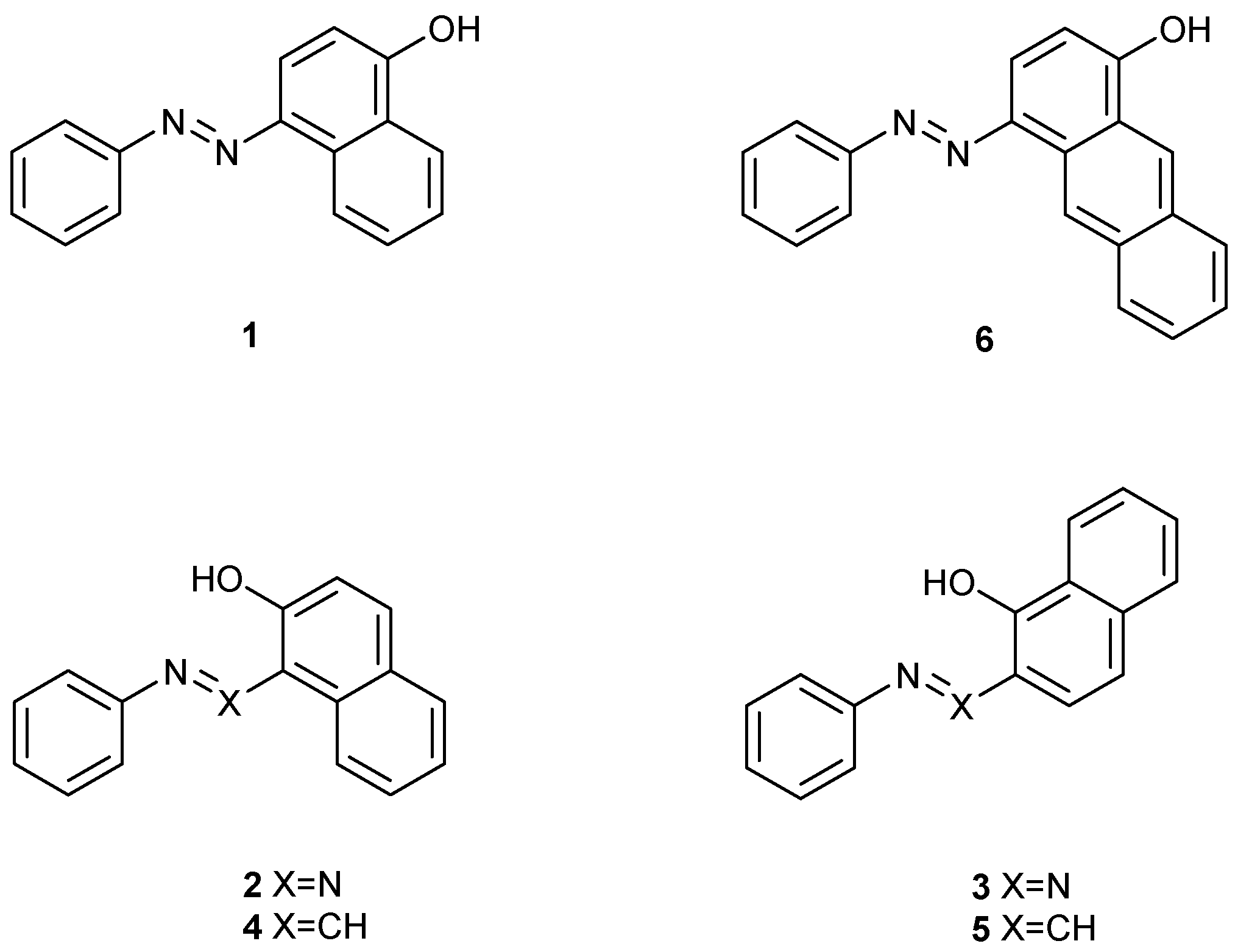{kind=link}
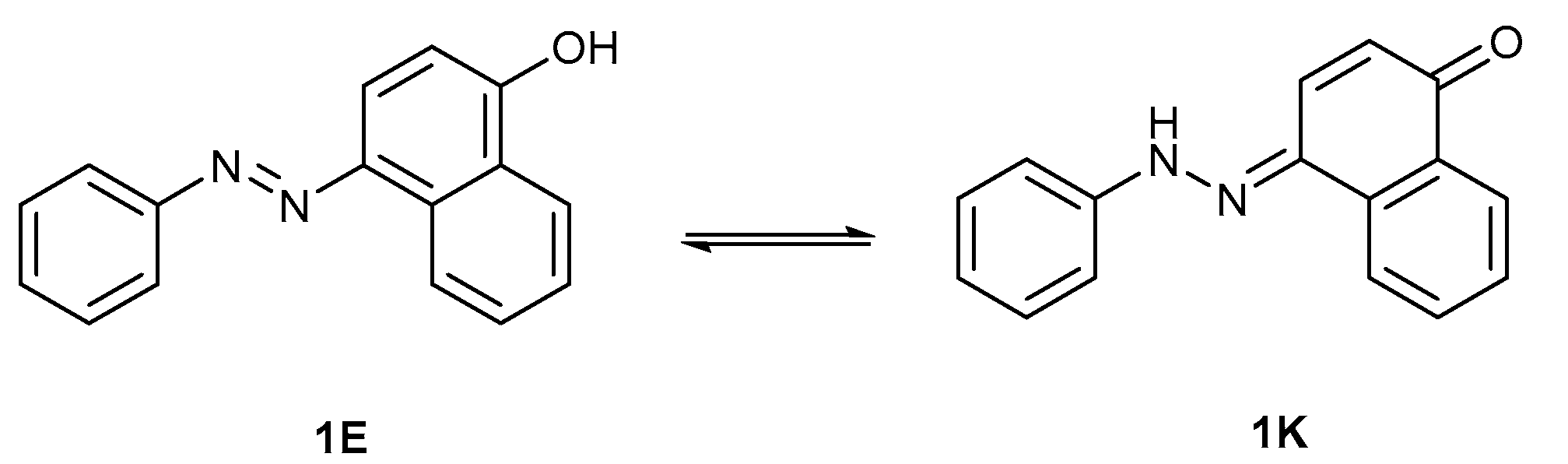{kind=link}
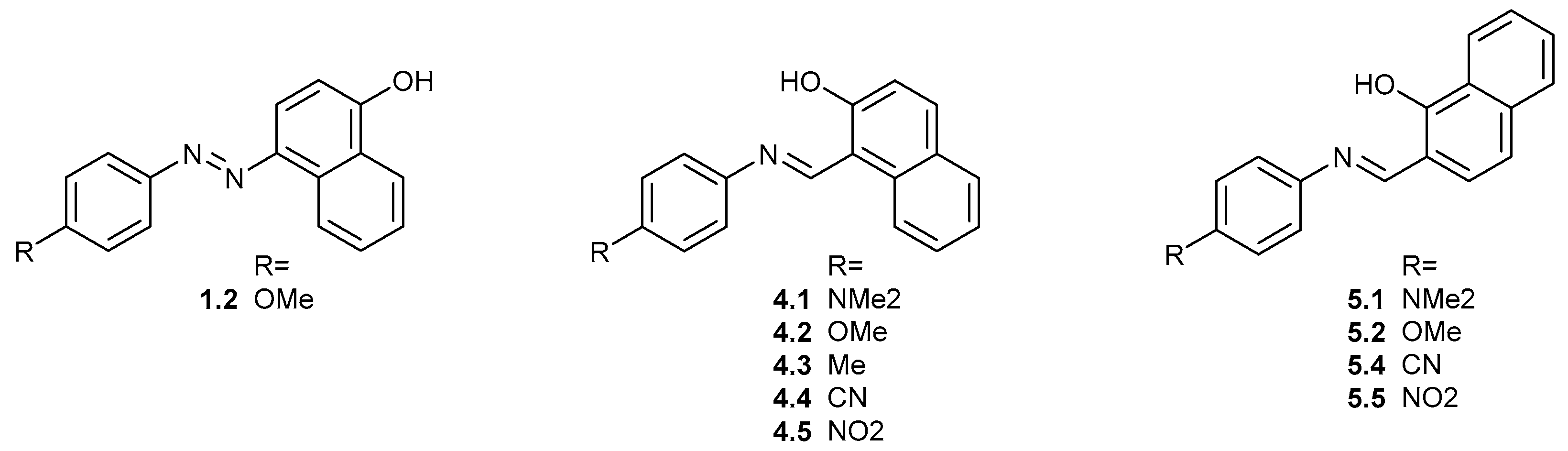{kind=link}
| Functional Name | ΔEpredicted kcal/mol | Linearity: ΔEpredicted = a. ΔGexperimental + b | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | a | b | R *** | |
| TautLYP | 1.49 | 0.39 | 0.22 | 1.54 | 0.80 | −1.35 | 1.04 | −0.01 | 0.996 |
| MN12-SX [42] | 2.00 | 0.48 | −0.35 | 1.58 | 0.79 | −0.60 | 0.91 | 0.25 | 0.936 |
| BHandH [43] | 1.01 | 0.22 | −0.42 | 1.50 | 0.68 | −2.15 | 1.22 | −0.39 | 0.988 |
| M06-2X [44] | 1.88 | 0.82 | 0.18 | 2.23 | 1.41 | −1.19 | 1.19 | 0.38 | 0.996 |
| HF [30,45] | 1.16 | 0.28 | −0.22 | 0.55 | −0.14 | −2.83 | 1.19 | −0.71 | 0.906 |
| SOGGA11-X [46] | 0.73 | −0.65 | −1.40 | 0.42 | −0.42 | −2.25 | 1.03 | −1.04 | 0.975 |
| M11 [47] | 0.83 | −0.89 | −1.34 | 0.51 | −0.13 | −2.59 | 1.19 | −1.11 | 0.986 |
| BHandHLYP [43] | 0.42 | −0.65 | −1.30 | 0.53 | −0.25 | −2.80 | 1.17 | −1.18 | 0.996 |
| CAM-B3LYP [48] | −0.44 | −1.15 | −1.71 | 0.31 | −0.41 | −3.58 | 1.27 | −1.71 | 0.978 |
| HISSbPBE [49] | −0.70 | −1.03 | −1.83 | 0.31 | −0.64 | −3.60 | 1.23 | −1.78 | 0.965 |
| ωB97X [50] | −0.15 | −1.39 | −1.88 | 0.07 | −0.62 | −3.59 | 1.28 | −1.81 | 0.995 |
| PBE0 [51] | −0.90 | −1.08 | −1.90 | 0.37 | −0.54 | −3.57 | 1.21 | −1.79 | 0.951 |
| PBEh1PBE [52] | −0.92 | −1.19 | −2.01 | 0.25 | −0.65 | −3.59 | 1.20 | −1.87 | 0.958 |
| APF [53] | −1.07 | −1.19 | −2.01 | 0.27 | −0.63 | −3.71 | 1.22 | −1.91 | 0.944 |
| ωB97X-D [54] | −0.60 | −1.41 | −2.00 | 0.08 | −0.66 | −3.81 | 1.29 | −1.96 | 0.984 |
| HSE06 [55] | −1.04 | −1.20 | −2.05 | 0.20 | −0.72 | −3.63 | 1.18 | −1.92 | 0.951 |
| LC-ωPBE [56] | −0.87 | −1.41 | −1.86 | 0.35 | −0.34 | −4.46 | 1.51 | −2.08 | 0.950 |
| APFD [53] | −1.45 | −1.29 | −2.11 | 0.26 | −0.62 | −4.07 | 1.28 | −2.10 | 0.914 |
| B3PW91 [57,58] | −1.34 | −1.33 | −2.17 | 0.14 | −0.77 | −3.91 | 1.22 | −2.09 | 0.932 |
| X3LYP [59] | −1.19 | −1.49 | −2.26 | −0.16 | −1.00 | −3.80 | 1.14 | −2.14 | 0.963 |
| MP2 [60] | 3.70 | 2.03 | 1.24 | 3.75 | 3.18 | 1.45 | 0.96 | 2.14 | 0.902 |
| B3LYP [61,62] | −1.36 | −1.58 | −2.37 | −0.24 | −1.09 | −3.93 | 1.15 | −2.26 | 0.957 |
| TPSSh [63] | −1.61 | −1.49 | −2.35 | −0.31 | −1.22 | −4.01 | 1.11 | −2.31 | 0.930 |
| τ-HCTHhyb [64] | −1.87 | −1.58 | −2.45 | −0.12 | −1.02 | −4.31 | 1.22 | −2.42 | 0.903 |
| N12SX [42] | −1.95 | −1.79 | −2.58 | −0.22 | −1.16 | −4.59 | 1.28 | −2.60 | 0.913 |
| TPSS [65] | −2.44 | −1.73 | −2.64 | −0.68 | −1.58 | −4.58 | 1.08 | −2.74 | 0.861 |
| O3LYP [66,67] | −2.44 | −2.06 | −2.90 | −0.48 | −1.43 | −4.85 | 1.25 | −2.90 | 0.889 |
| BMK [68] | −3.01 | −3.16 | −3.94 | −0.74 | −1.65 | −5.99 | 1.57 | −3.76 | 0.906 |
| M06-HF [44] | 6.34 | 4.92 | 4.55 | 4.00 | 3.81 | 2.72 | 0.70 | 4.09 | 0.609 |
| experiment **** | 1.35 | 0.42 | −0.24 | 1.42 | 0.94 | −1.31 | |||
| Functional Name | Type * | HF Exchange [%] | MAE ** | MAPE *** [%] | MSE **** | MSPE ***** [%] |
|---|---|---|---|---|---|---|
| TautLYP | GH-GGA | 43.79 | 0.08 | 8.4 | 0.01 | 84.6 |
| MN12-SX | RSH-mGGA | 25–0 | 0.31 | 31.5 | 0.16 | 1326 |
| BHandH | GH-GGA | 50 | 0.32 | 41.5 | 0.16 | 2336 |
| M06-2X | GH-mGGA | 54 | 0.46 | 71.5 | 0.25 | 8019 |
| HF | 100 | 0.64 | 58.1 | 0.72 | 5307 | |
| SOGGA11-X | GH-GGA | 40.15 | 1.03 | 179 | 1.11 | 56,652 |
| M11 | RSH-mGGA | 42.8–100 | 1.03 | 182 | 1.14 | 57,162 |
| BHandHLYP | GH-GGA | 50 | 1.11 | 179 | 1.26 | 50,795 |
| CAM-B3LYP | RSH-GGA | 19–65 | 1.59 | 254 | 2.61 | 100,203 |
| HISSbPBE | RSH-GGA | 0–60–0 | 1.68 | 265 | 2.97 | 109,721 |
| ωB97X | RSH-GGA | 15.77–100 | 1.69 | 278 | 2.95 | 124,477 |
| PBE0 | GH-GGA | 25 | 1.70 | 271 | 3.08 | 118,265 |
| PBEh1PBE | GH-GGA | 25 | 1.78 | 287 | 3.34 | 133,408 |
| APF | GH-GGA | 23 | 1.82 | 290 | 3.53 | 134,506 |
| ωB97X-D | RSH-GGA + D | 22.2–100 | 1.83 | 296 | 3.48 | 139,486 |
| HSE06 | RSH-GGA | 25–0 | 1.84 | 294 | 3.54 | 139,086 |
| LC-ωPBE | RSH-GGA | 0–100 | 1.86 | 289 | 3.94 | 127,711 |
| APFD | GH-GGA + D | 23 | 1.98 | 310 | 4.29 | 152,067 |
| B3PW91 | GH-GGA | 20 | 1.99 | 316 | 4.23 | 159,313 |
| X3LYP | GH-GGA | 21.8 | 2.08 | 333 | 4.44 | 176,504 |
| MP2 | 100 | 2.13 | 298 | 4.72 | 115,308 | |
| B3LYP | GH-GGA | 20 | 2.19 | 352 | 4.95 | 196,692 |
| TPSSh | GH-mGGA | 10 | 2.26 | 353 | 5.31 | 193,046 |
| τ-HCTHhyb | GH-mGGA | 15 | 2.32 | 365 | 5.75 | 210,342 |
| N12SX | RSH-GGA | 25–0 | 2.48 | 391 | 6.52 | 239,972 |
| TPSS | mGGA | 0 | 2.71 | 411 | 7.72 | 253,502 |
| O3LYP | GH-GGA | 11.61 | 2.79 | 442 | 8.23 | 307,829 |
| BMK | mGGA | 42 | 3.51 | 586 | 13.1 | 582,436 |
| M06-HF | GH-mGGA | 100 | 3.96 | 709 | 16.5 | 930,805 |
| Functional Name | MAPE [%] | MAE | Non-Planarity Description * | |||
|---|---|---|---|---|---|---|
| Bonds | Angles | Total | Bonds [Å] | Angles [o] | ||
| TautLYP | 3.31 | 42.27 | 45.57 | 0.05 | 3.81 | yes |
| MN12-SX | 2.95 | 199.70 | 202.65 | 0.04 | 20.72 | no |
| BHandH | 4.95 | 199.99 | 204.93 | 0.07 | 20.75 | no |
| M06-2X | 2.61 | 114.56 | 117.17 | 0.04 | 12.26 | yes |
| HF | 4.53 | 48.04 | 52.57 | 0.06 | 4.34 | yes |
| SOGGA11-X | 2.76 | 140.41 | 143.16 | 0.04 | 14.91 | no (1), yes (6) |
| M11 | 2.55 | 200.00 | 202.55 | 0.03 | 20.75 | no |
| BHandHLYP | 3.38 | 141.55 | 144.93 | 0.05 | 15.00 | no (1), yes (6) |
| Cmpd | R | Solvent | |
|---|---|---|---|
| CCl4 | CH3CN | ||
| 1 | H | −0.03 | |
| 1.2 | OCH3 | 1.49 | |
| 2 | H | 0.34 | −0.15 |
| 3 | H | −0.39 | −0.47 |
| 4 | H | 1.40 | 0.30 |
| 4.1 | N(CH3)2 | 1.33 | 0.37 |
| 4.2 | OCH3 | 1.34 | 0.37 |
| 4.3 | CH3 | 1.33 | 0.23 |
| 4.4 | CN | 1.42 | 0.55 |
| 4.5 | NO2 | 1.48 | 0.47 |
| 5 | H | 0.79 | −0.34 |
| 5.1 | N(CH3)2 | 1.12 | −0.45 |
| 5.2 | OCH3 | 0.98 | −0.20 |
| 5.4 | CN | 0.91 | −0.10 |
| 5.5 | NO2 | 1.00 | −0.15 |
| Functional | MAE | MAPE [%] | MAPE [%] C6H12, CCl4, CH3CN | ||
|---|---|---|---|---|---|
| CCl4 | CH3CN | CCl4 | CH3CN | ||
| TautLYP | 0.16 | 0.09 | 14.9 | 62.3 | 85.7 |
| MN12-SX | 0.18 | 0.18 | 15.8 | 97.3 | 144 |
| BHandH | 0.17 | 0.14 | 18.0 | 182 | 242 |
| M06-2X | 0.72 | 0.67 | 74.5 | 335 | 481 |
| B3LYP | 1.78 | 2.08 | 216 | 1401 | 1969 |
| ΔEpredicted kcal/mol | Linearity: ΔEpredicted = a. ΔGexperiment + b | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | a | b | R | |
| TautLYP | 2.49 | 0.80 | 0.09 | 2.34 | 1.57 | −0.42 | 1.09 | 0.67 | 0.965 |
| MP2 | 4.41 | 2.27 | 1.41 | 4.43 | 3.82 | 2.09 | 1.04 | 2.62 | 0.837 |
| TautLYP * | 2.70 | 0.63 | −0.14 | 2.38 | 1.62 | −0.21 | 1.10 | 0.69 | 0.922 |
| MP3 * | 4.03 | 2.64 | 2.11 | 4.56 | 4.00 | 1.11 | 1.24 | 2.54 | 0.976 |
| MP4 * | 1.67 | 0.41 | −0.49 | 2.51 | 1.84 | −0.48 | 1.10 | 0.44 | 0.909 |
| CCSD(T) * | 2.77 | 1.53 | 0.78 | 3.40 | 2.78 | 0.40 | 1.10 | 1.47 | 0.949 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antonov, L. Tautomerism in Azo and Azomethyne Dyes: When and If Theory Meets Experiment. Molecules 2019, 24, 2252. https://doi.org/10.3390/molecules24122252
Antonov L. Tautomerism in Azo and Azomethyne Dyes: When and If Theory Meets Experiment. Molecules. 2019; 24(12):2252. https://doi.org/10.3390/molecules24122252
Chicago/Turabian StyleAntonov, Liudmil. 2019. "Tautomerism in Azo and Azomethyne Dyes: When and If Theory Meets Experiment" Molecules 24, no. 12: 2252. https://doi.org/10.3390/molecules24122252
APA StyleAntonov, L. (2019). Tautomerism in Azo and Azomethyne Dyes: When and If Theory Meets Experiment. Molecules, 24(12), 2252. https://doi.org/10.3390/molecules24122252