
Merging Ligand-Based and Structure-Based Methods in Drug Discovery: An Overview of Combined Virtual Screening Approaches


Abstract
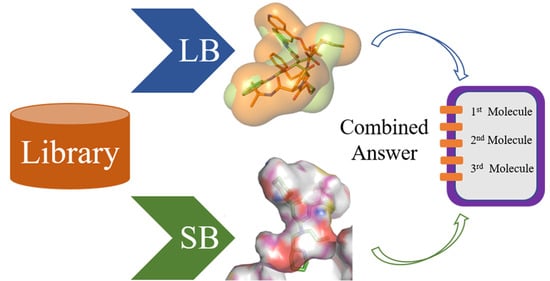
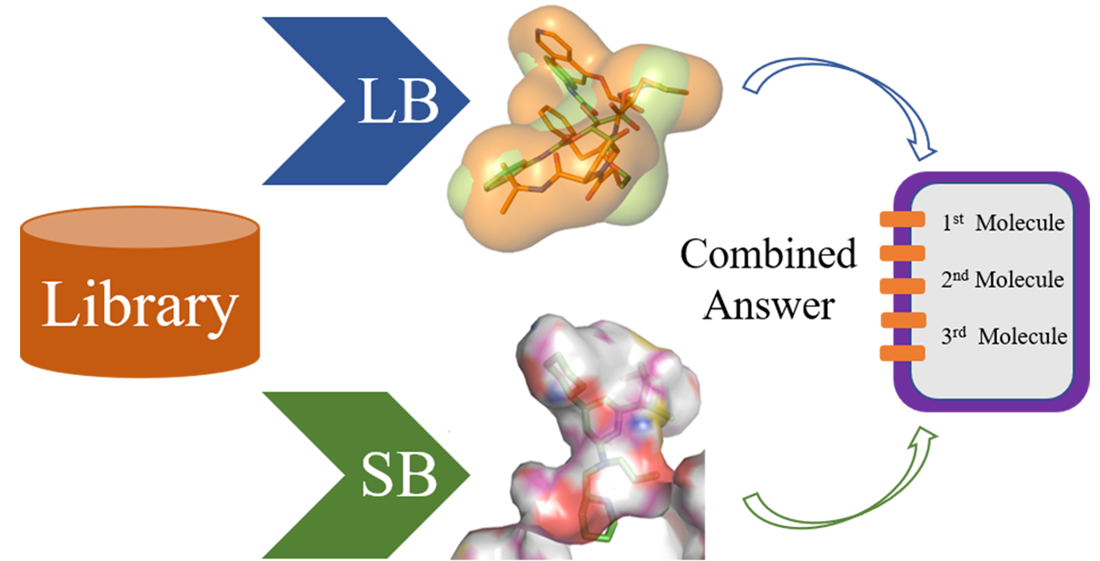
:
1. Introduction
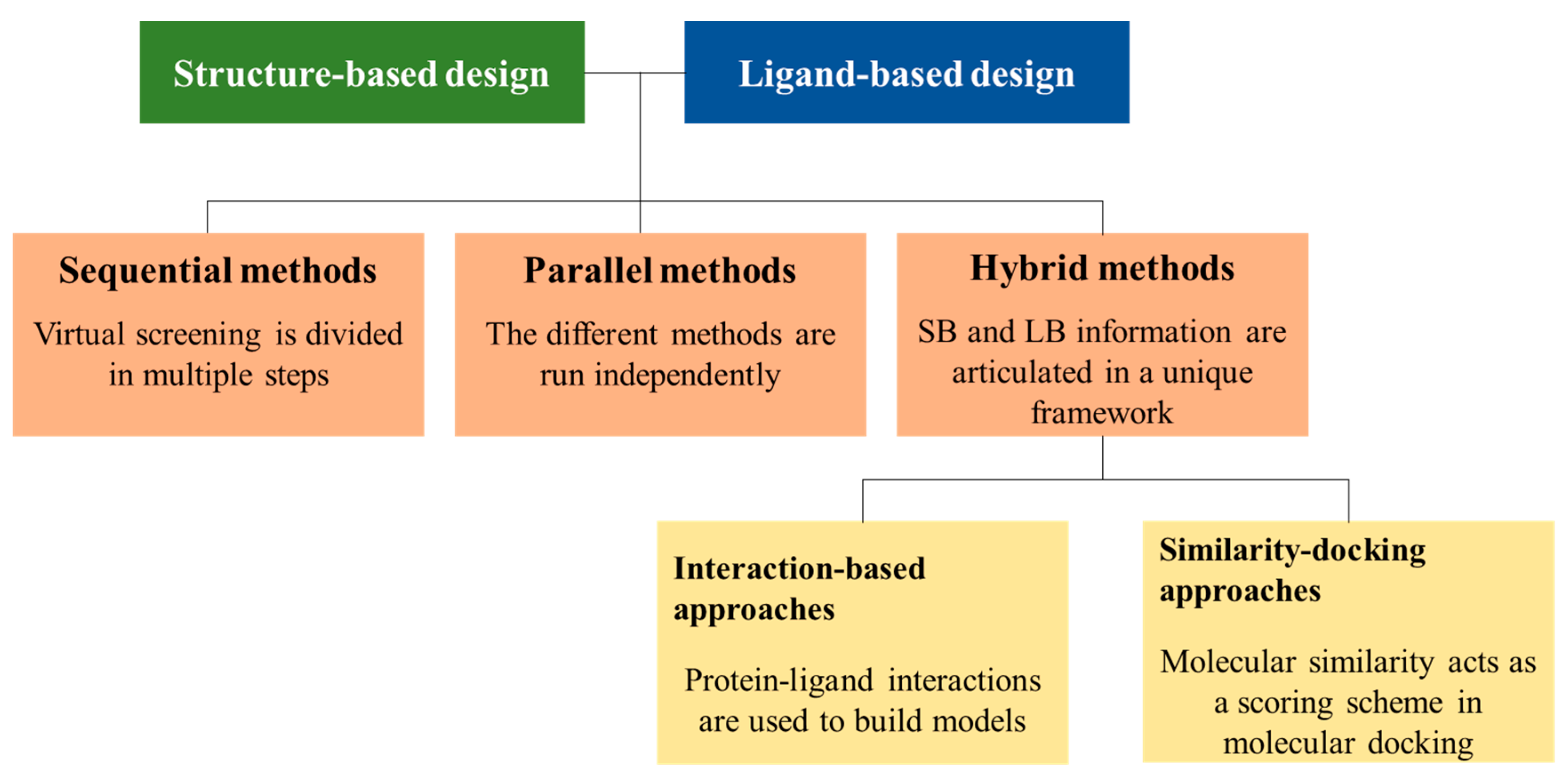
2. LB and SB Strategies in VS
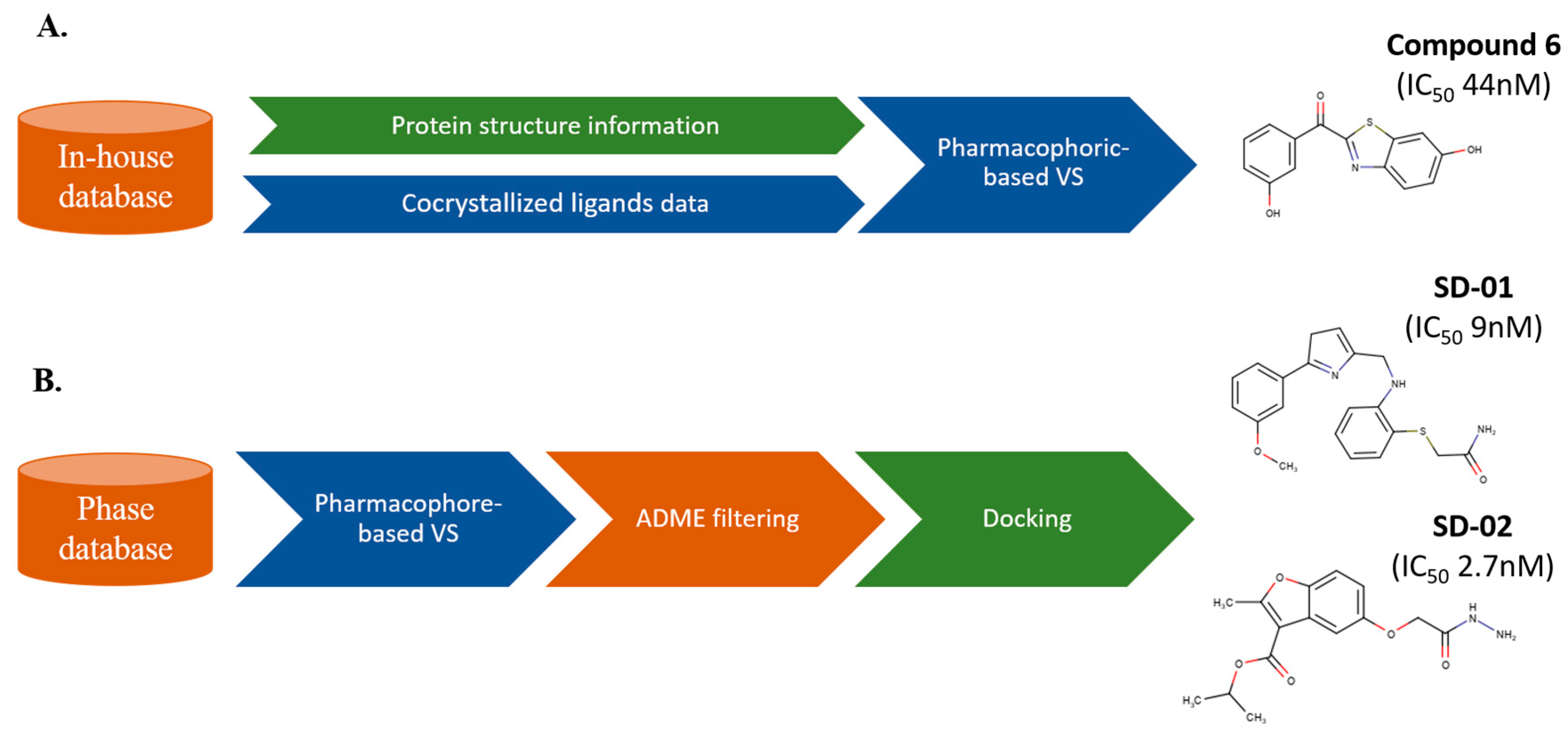
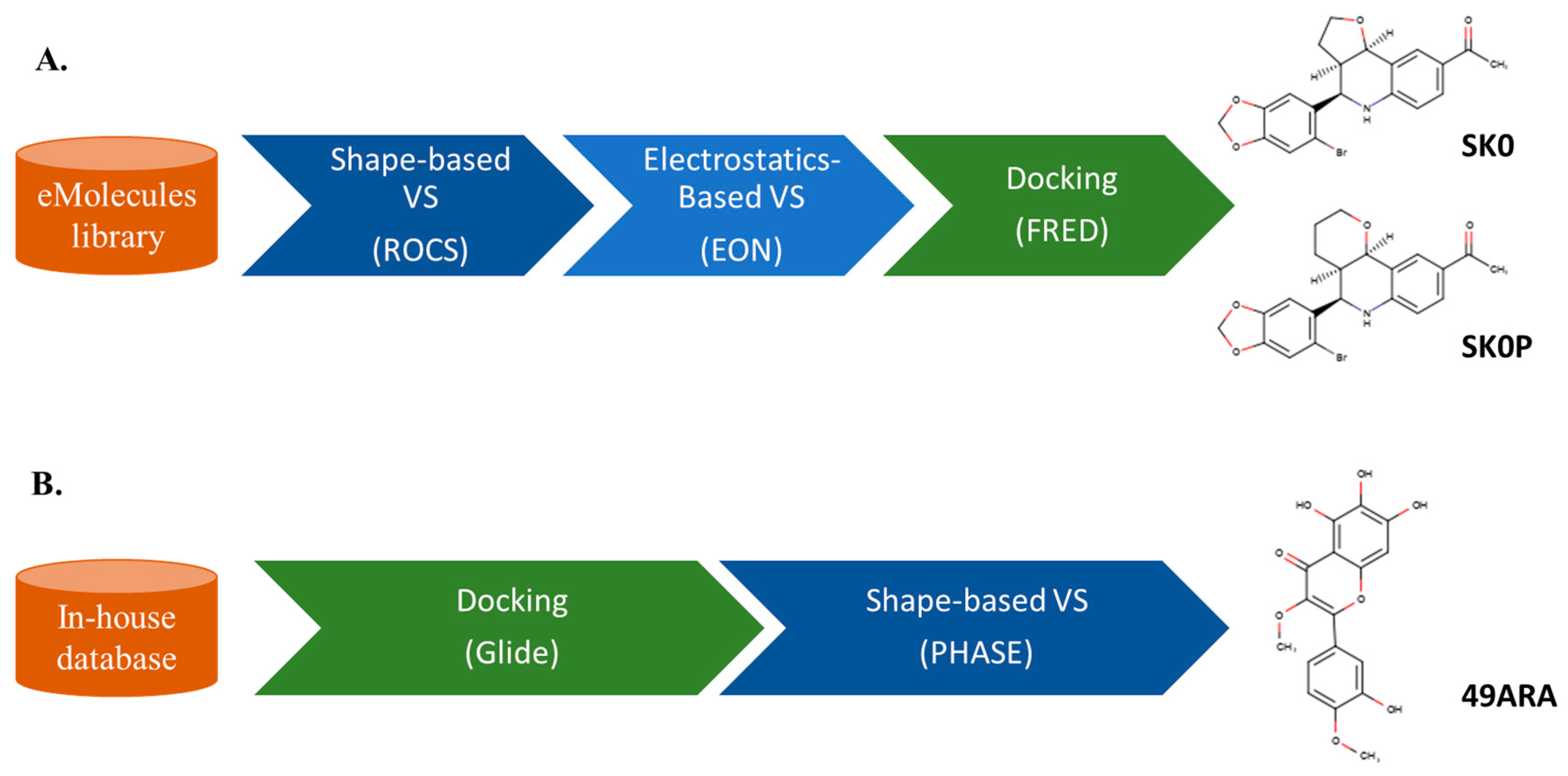
3. Sequential LB and SB Methods
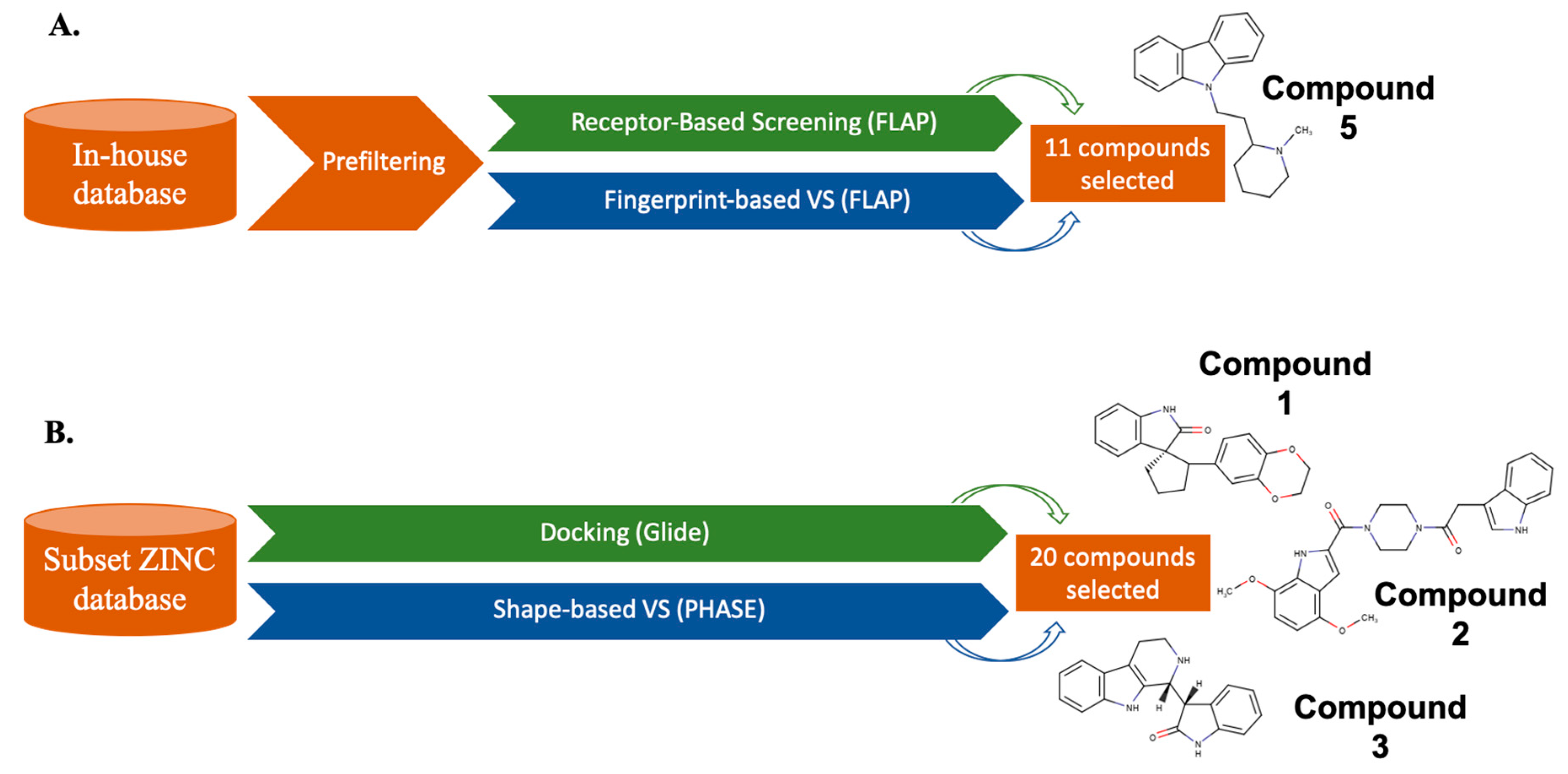
4. Parallel LB and SB Approaches
5. Hybrid Approaches
5.1. Interaction-Based Methods
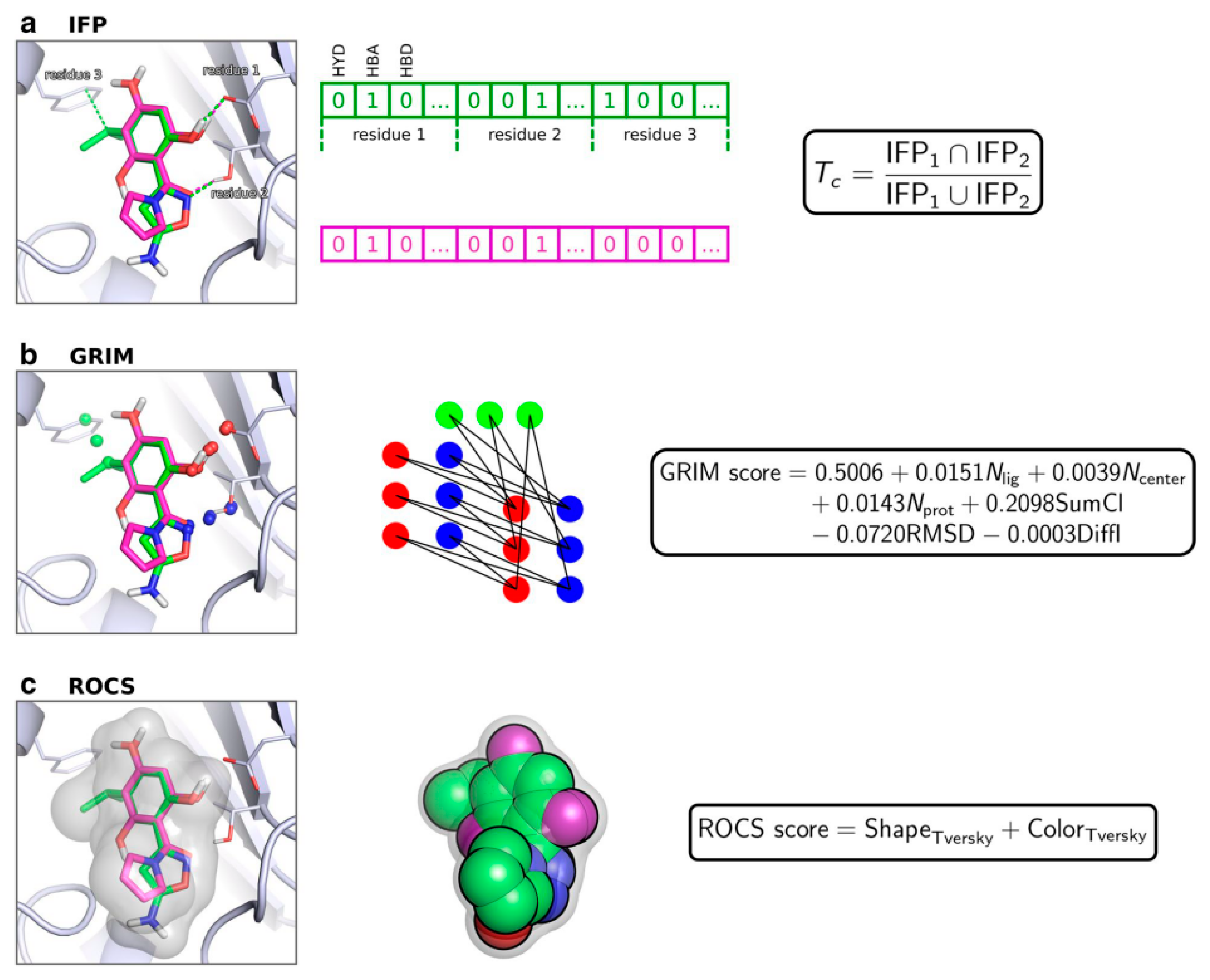
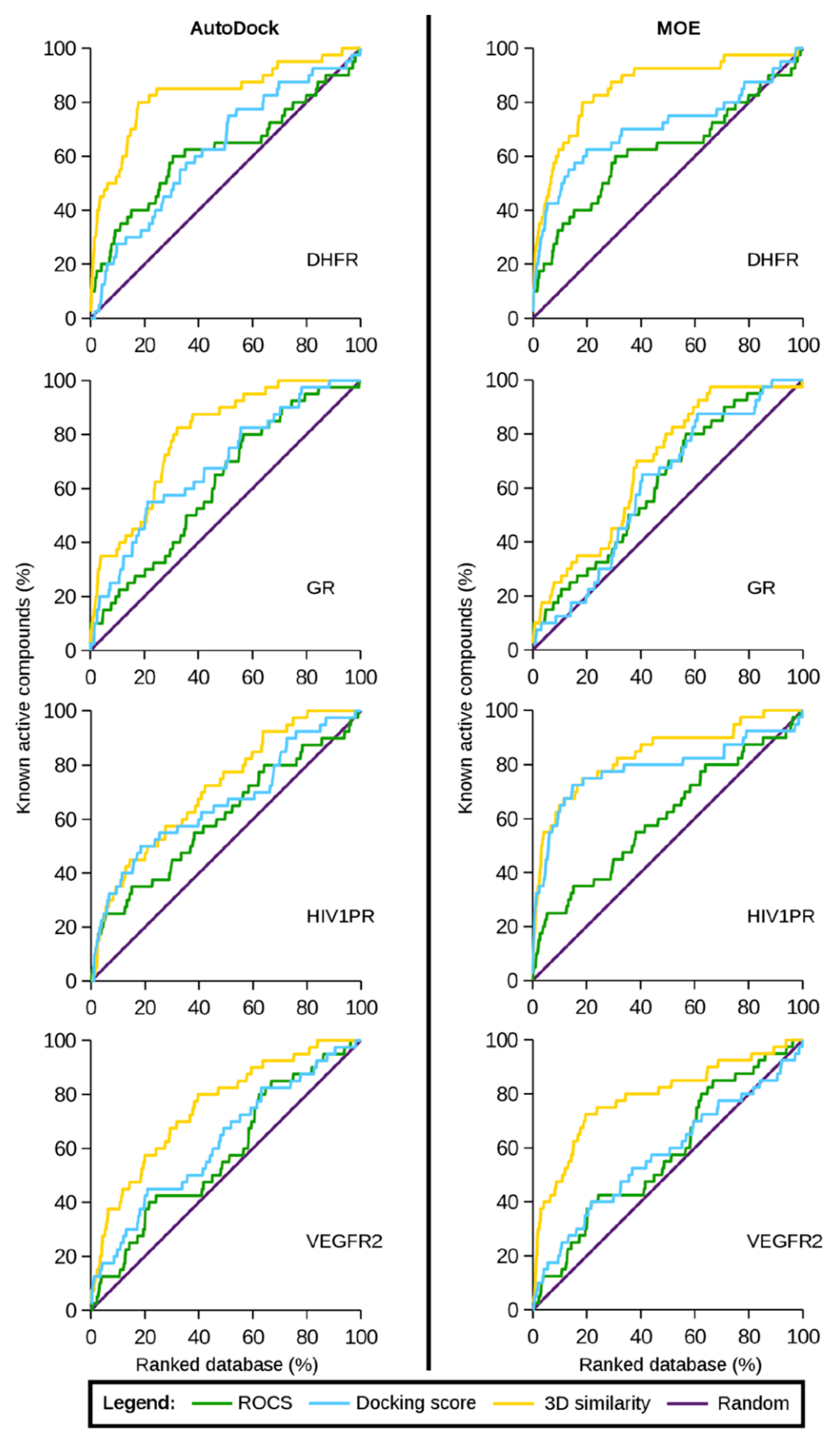
5.2. Similarity-Docking Strategies
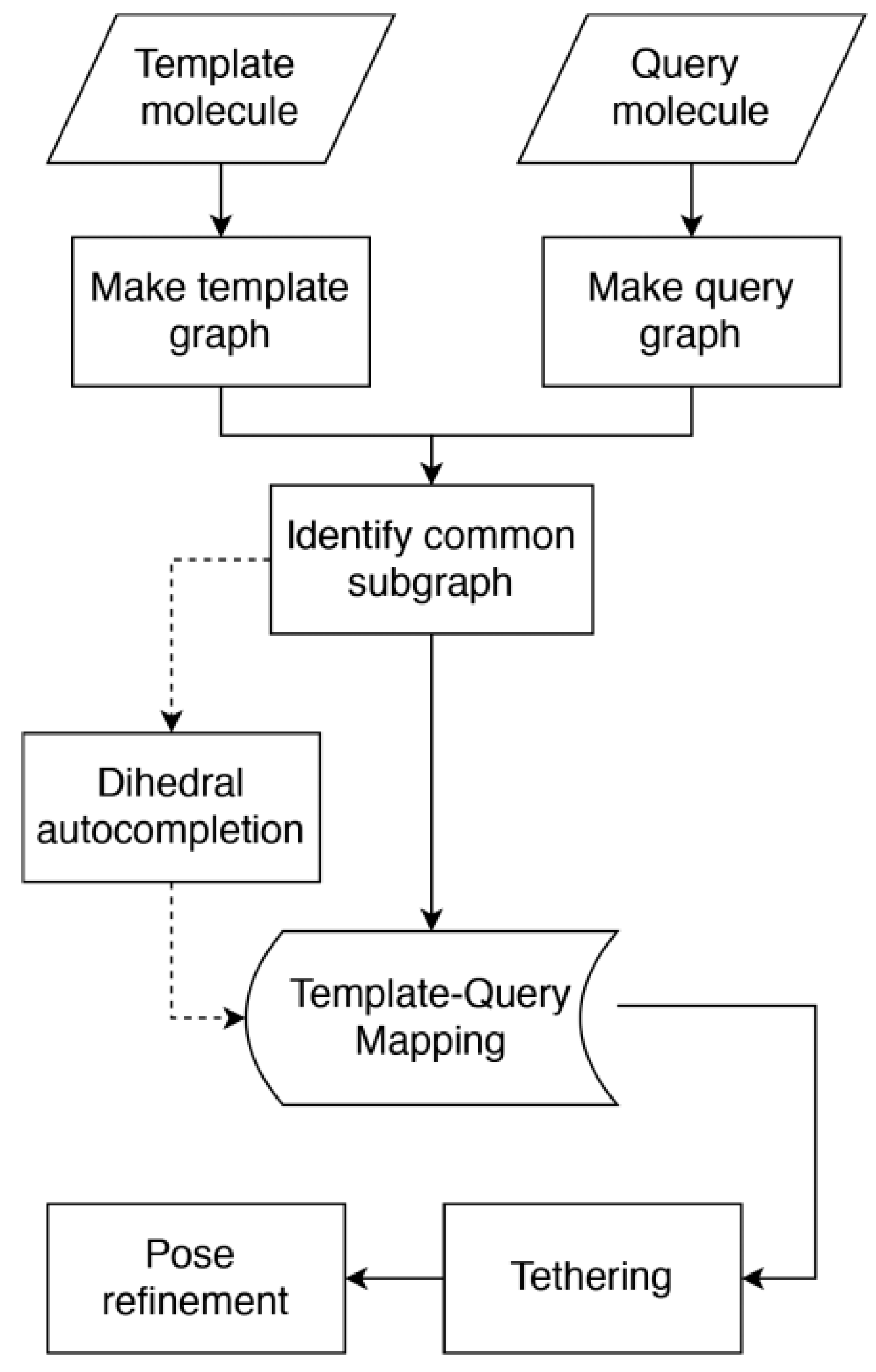
5.2.1. Predicting the Pose of Ligands
5.2.2. Similarity-Guided Score Scheme
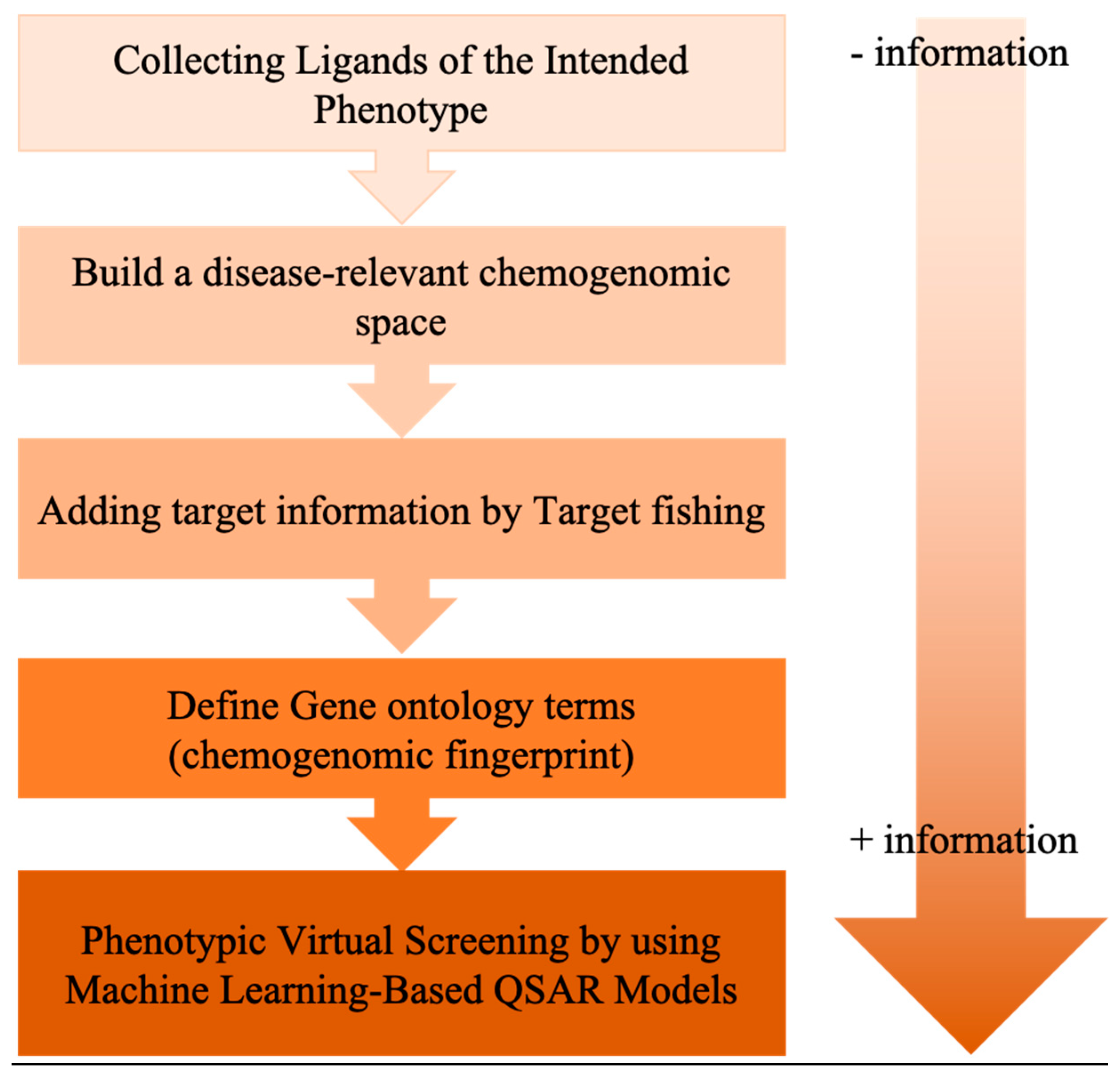
6. Exploiting Chemical Libraries and Biological Data
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Free Energy Calculations. Theory and Applications in Chemistry and Biology; Chipot, C., Pohorille, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Abel, R.; Wang, L.; Harder, E.D.; Berne, B.J.; Friesner, R.A. Advancing drug discovery through enhanced free energy calculations. Acc. Chem. Res. 2017, 50, 1625–1632. [Google Scholar] [CrossRef] [PubMed]
- Williams-Noonan, B.J.; Yuriev, E.; Chalmers, D.K. Free energy methods in drug design: Prospects of “alchemical perturbation” in medicinal chemistry. J. Med. Chem. 2018, 61, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Christ, C.D.; Fox, T. Accuracy assessment and automation of free energy calculations for drug design. J. Chem. Inf. Model. 2013, 54, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Mondal, D.; Florian, J.; Warshel, A. Exploring the effectiveness of binding free energy calculations. J. Phys. Chem. B 2019, 123, 8910–8915. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Allen, B.; Sherman, W. Relative binding free energy calculations in drug discovery: Recent advances and practical considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef]
- Zhang, H.; Gattuso, H.; Dumont, E.; Cai, W.; Monari, A.; Chipot, C.; Dehez, F. Accurate estimation of the standard binding free energy of netropsin with DNA. Molecules 2018, 23, 228. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Gumbart, J.C.; Chen, H.; Shao, X.; Cai, W.; Chipot, C. BFFE: A user-friendly graphical interface facilitating absolute binding free-energy calculations. J. Chem. Inf. Model. 2018, 58, 556–560. [Google Scholar] [CrossRef]
- Jespers, W.; Esguerra, M.; Aqvist, J.; Gutiérrez-de-Terán, H.J. QligFEP: An automated workflow for small molecule free energy calculations in Q. J. Cheminform. 2019, 11, 26. [Google Scholar] [CrossRef]
- Gioia, D.; Bertazzo, M.; Recanatini, M.; Masetti, M.; Cavalli, A. Dynamic docking: A paradigm shift in computational drug discovery. Molecules 2017, 22, 2029. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Carmona, S.; Schmidtke, P.; Luque, F.J.; Baker, L.; Matassova, N.; Davis, B.; Roughley, S.; Murray, J.; Hubbard, R.; Barril, X. Dynamic undocking and the quasi-bound state as tools for drug discovery. Nat. Chem. 2017, 9, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Cavasotto, C.N.; Aucar, M.G. High-throughput docking using quantum mechanical scoring. Front. Chem. 2020, 8, 246. [Google Scholar] [CrossRef] [PubMed]
- Colizzi, F.; Perozzo, R.; Scapozza, L.; Recanatini, M.; Cavalli, A. Single-molecule pulling simulations can discern active from inactive enzyme inhibitors. J. Am. Chem. Soc. 2010, 132, 7361–7371. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; García-Vallvé, S. The light and dark sides of virtual screening: What is there to know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuo, N.; Sekijima, M. Improved method of structure-based virtual screening via interaction-energy-based learning. J. Chem. Inf. Model. 2019, 59, 1050–1061. [Google Scholar] [CrossRef] [Green Version]
- Ertl, P. Cheminformatics analysis of organic substituents: Identification of the most common substituents, calculation of substituent properties, and automatic identification of drug-like bioisosteric groups. J. Chem. Inf. Comput. Sci. 2003, 43, 374–380. [Google Scholar] [CrossRef]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef]
- Schneider, G. Virtual screening: An endless staircase? Nat. Rev. Drug Discov. 2010, 9, 273–276. [Google Scholar] [CrossRef]
- Westermaier, Y.; Barril, X.; Scapozza, L. Virtual screening: An in silico tool for interlacing the chemical universe with the proteome. Methods 2015, 71, 44–57. [Google Scholar] [CrossRef]
- Taylor, R.D.; Jewsbury, P.J.; Essex, J.W. A review of protein-small molecule docking methods. J. Comput. Aided Mol. Des. 2002, 16, 151–166. [Google Scholar] [CrossRef]
- Li, J.; Fu, A.; Zhang, L. An overview of scoring functions used for protein–ligand interactions in molecular docking. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef]
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva, F.P., Jr. Key topics in molecular docking for drug design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Concepts and Applications of Molecular Similarity; Johnson, M.A.; Maggiora, G.M. (Eds.) John Wiley & Sons: New York, NY, USA, 1990. [Google Scholar]
- Jørgensen, A.M.M.; Pedersen, J.T. Structural diversity of small molecule libraries. J. Chem. Inf. Comput. Sci. 2001, 41, 338–345. [Google Scholar] [CrossRef]
- Ivanciuc, O.; Taraviras, S.L.; Cabrol-Bass, D. Quasi-orthogonal basis sets of molecular graph descriptors as a chemical diversity measure. J. Chem. Inf. Comput. Sci. 2000, 40, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 2010, 29, 157–170. [Google Scholar] [CrossRef]
- Cross, S.; Baroni, M.; Carosati, E.; Benedetti, P.; Clementi, S. FLAP: GRID Molecular interaction fields in virtual screening. Validation using the DUD data set. J. Chem. Inf. Model. 2010, 50, 1442–1450. [Google Scholar] [CrossRef]
- Mestres, J.; Rohrer, D.C.; Maggiora, G.M. MIMIC: A molecular-field matching program. Exploiting applicability of molecular similarity approaches. J. Comput. Chem. 1997, 18, 934–954. [Google Scholar] [CrossRef]
- Cheeseright, T.J.; Mackey, M.D.; Melville, J.L.; Vinter, J.G. FieldScreen: Virtual screening using molecular fields. Application to the DUD data set. J. Chem. Inf. Model. 2008, 48, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, J.; Deplano, A.; Herrero, A.; Ginex, T.; Gibert, E.; Rabal, O.; Oyarzabal, J.; Herrero, E.; Luque, F.J. Development and validation of molecular overlays derived from three-dimensional hydrophobic similarity with PharmScreen. J. Chem. Inf. Model. 2018, 58, 1596–1609. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef]
- Sastry, G.M.; Dixon, S.L.; Sherman, W. Rapid shape-based ligand alignment and virtual screening method based on atom/feature-pair similarities and volume overlap scoring. J. Chem. Inf. Model. 2011, 51, 2455–2466. [Google Scholar] [CrossRef] [PubMed]
- Abrahamian, E.; Fox, P.C.; Nærum, L.; Thøger Christensen, I.; Thøgersen, H.; Clark, R.D. Efficient generation, storage, and manipulation of fully flexible pharmacophore multiplets and their use in 3-D similarity searching. J. Chem. Inf. Comput. Sci. 2003, 43, 458–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperandio, O.; Miteva, M.; Villoutreix, B. Combining ligand- and structure-based methods in drug design projects. Curr. Comput. Aided Drug Des. 2008, 4, 250–258. [Google Scholar] [CrossRef]
- Talevi, A.; Gavernet, L.; Bruno-Blanch, L. Combined virtual screening strategies. Curr. Comput. Aided Drug Des. 2009, 5, 23–37. [Google Scholar] [CrossRef]
- Spadaro, A.; Negri, M.; Marchais-Oberwinkler, S.; Bey, E.; Frotscher, M. Hydroxybenzothiazoles as new nonsteroidal inhibitors of 17®-hydroxysteroid dehydrogenase type 1 (17®-HSD1). PLoS ONE 2012, 7, 29252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debnath, S.; Debnath, T.; Bhaumik, S.; Majumdar, S.; Kalle, A.M.; Aparna, V. Discovery of novel potential selective HDAC8 inhibitors by combine ligand-based, structure-based virtual screening and in-vitro biological evaluation. Sci. Rep. 2019, 9, 17174. [Google Scholar] [CrossRef]
- Wilson, G.L.; Lill, M.A. Integrating structure-based and ligand-based approaches for computational drug design. Future Med. Chem. 2011, 3, 735–750. [Google Scholar] [CrossRef]
- Drwal, M.N.; Griffith, R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov. Today Technol. 2013, 10, e395–e401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, H.; Shen, C.; Hu, X.; Gao, J.; Li, D.; Cao, D.; Hou, T. Combined strategies in structure-based virtual screening. Phys. Chem. Chem. Phys. 2020, 22, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Spyrakis, F.; Bidon-Chanal, A.; Barril, X.; Luque, F.J. Protein flexibility and ligand recognition: Challenges for molecular modeling. Curr. Top. Med. Chem. 2011, 11, 192–210. [Google Scholar] [CrossRef]
- Lexa, K.W.; Carlson, H.A. Protein flexibility in docking and surface mapping. Q. Rev. Biophys. 2012, 45, 301–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmaso, V.; Moro, S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: An overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, A.; Ross, G.A.; Biggin, P.C. Waterdock 2.0: Water placement prediction for Holo-structures with a Pymol plugin. PLoS ONE 2017, 12, e0172743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudling, A.; Orro, A.; Carlsson, J. Prediction of ordered water molecules in protein binding sites from molecular dynamics simulations: The impact of ligand binding on hydration networks. J. Chem. Inf. Model. 2018, 58, 350–361. [Google Scholar] [CrossRef] [Green Version]
- Sciebel, J.; Gaspari, R.; Wulsdorf, T.; Ngo, K.; Sohn, C.; Schrader, T.E.; Cavalli, A.; Ostermann, A.; Heine, A.; Klebe, G. Intriguing role of water in protein-ligand binding studies by neutro crystallography on trypsin complexes. Nat. Commun. 2018, 9, 3559. [Google Scholar] [CrossRef] [Green Version]
- Maurer, M.; Oostenbrink, C. Water in protein hydration and ligand recognition. J. Mol. Recog. 2019, 32, e2810. [Google Scholar] [CrossRef] [PubMed]
- Geschwindner, S.; Ulander, J. The current impact of water thermodynamics for small-molecule drug discovery. Expert Opin. Drug Discov. 2019, 14, 1221–1225. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Liu, J.; Wang, R. Classification of current scoring functions. J. Chem. Inf. Model. 2015, 55, 475–482. [Google Scholar] [CrossRef]
- Guedes, I.A.; Pereira, F.S.S.; Dardenne, L.E. Empirical scoring functions for structure-based virtual screening: Applications, critical aspects, and challenges. Front. Pharmacol. 2018, 9, 1089. [Google Scholar] [CrossRef] [PubMed]
- Palacio-Rodríguez, K.; Lans, I.; Cavasotto, C.N.; Cossio, P. Exponential consensus ranking improves the outcome in docking and receptor ensemble docking. Sci. Rep. 2019, 9, 5142. [Google Scholar] [CrossRef] [PubMed]
- Hein, M.; Zilian, D.; Sotriffer, C.A. Docking compared to 3D-pharmacophores: The scoring function challenge. Drug Discov. Today Technol. 2010, 4, e229–e236. [Google Scholar] [CrossRef]
- Eckert, H.; Bajorath, J. Molecular similarity analysis in virtual screening: Foundations, limitations and novel approaches. Drug Discov. Today 2007, 12, 225–233. [Google Scholar] [CrossRef]
- Grinter, S.Z.; Zou, X. Challenges, applications, and recent advances of protein-ligand docking in structure-based drug design. Molecules 2014, 19, 10150–10176. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Han, L.; Liu, Z.; Wang, R. Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results. J. Chem. Inf. Model. 2014, 54, 1717–1736. [Google Scholar] [CrossRef]
- Antunes, D.A.; Devaurs, D.; Kavraki, L.E. Understanding the challenges of protein flexibility in drug design. Expert Opin. Drug Discov. 2015, 10, 1301–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Takeda, S.; Kaneko, H.; Funatsu, K. Chemical-space-based de novo design method to generate drug-like molecules. J. Chem. Inf. Model. 2016, 56, 1885–1893. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [Green Version]
- Fischer, T.; Gazzola, S.; Riedl, R. Approaching target selectivity by de novo drug design. Expert. Opin. Drug Discov. 2019, 14, 791–803. [Google Scholar] [CrossRef]
- Méndez-Lucio, O.; Baillif, B.; Clevert, D.-A.; Rouquié, D.; Wichard, J. De novo generation of hit-like molecules from gene expression signatures using artificial intelligence. Nat. Commun. 2020, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Pei, J.; Lai, L. LigBuilder V3: A multi-target de novo drug design approach. Front. Chem. 2020, 8, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swann, S.L.; Brown, S.P.; Muchmore, S.W.; Patel, H.; Merta, P.; Locklear, J.; Hajduk, P.J. A Unified, probabilistic framework for structure- and ligand-based virtual screening. J. Med. Chem. 2011, 54, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Cleves, A.E.; Jain, A.N. Structure- and ligand-based virtual screening on DUD-E+: Performance dependence on approximations to the binding pocket. J. Chem. Inf. Model. 2020, 60, 4296–4310. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Vischer, H.F.; McNaught-Flores, D.; Leurs, R.; De Esch, I.J.P.; De Graaf, C. Function-specific virtual screening for GPCR ligands using a combined scoring method. Sci. Rep. 2016, 6, 28288. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Lounkine, E.; Bajorath, J. Similarity searching using fingerprints of molecular fragments involved in protein-ligand interactions. J. Chem. Inf. Model. 2008, 48, 2308–2312. [Google Scholar] [CrossRef]
- Tan, L.; Bajorath, J. Utilizing target-ligand interaction information in fingerprint searching for ligands of related targets. Chem. Biol. Drug Des. 2009, 74, 25–32. [Google Scholar] [CrossRef]
- Meslamani, J.; Li, J.; Sutter, J.; Stevens, A.; Bertrand, H.O.; Rognan, D. Protein-ligand-based pharmacophores: Generation and utility assessment in computational ligand profiling. J. Chem. Inf. Model. 2012, 52, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Fraccalvieri, D.; Andersson, C.D.; Bonati, L.; Linusson, A.; Andersson, P.L. Identification of potential aryl hydrocarbon receptor ligands by virtual screening of industrial chemicals. Environ. Sci. Pollut. Res. 2018, 25, 2436–2449. [Google Scholar] [CrossRef] [Green Version]
- Tanrikulu, Y.; Schneider, G. Pseudoreceptor models in drug design: Bridging ligand- and receptor-based virtual screening. Nat. Rev. Drug Discov. 2008, 7, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, D.G.; Buenemann, C.L.; Todorov, N.P.; Manallack, D.T.; Dean, P.M. Scaffold hopping in de novo design. Ligand generation in the absence of receptor information. J. Med. Chem. 2004, 47, 493–496. [Google Scholar] [CrossRef]
- Lorenzo, V.P.; Barbosa Filho, J.M.; Scotti, L.; Scotti, M.T. Combined structure- and ligand-based virtual screening to evaluate caulerpin analogs with potential inhibitory activity against monoamine oxidase B. Rev. Bras. Farmacogn. 2015, 25, 690–697. [Google Scholar] [CrossRef]
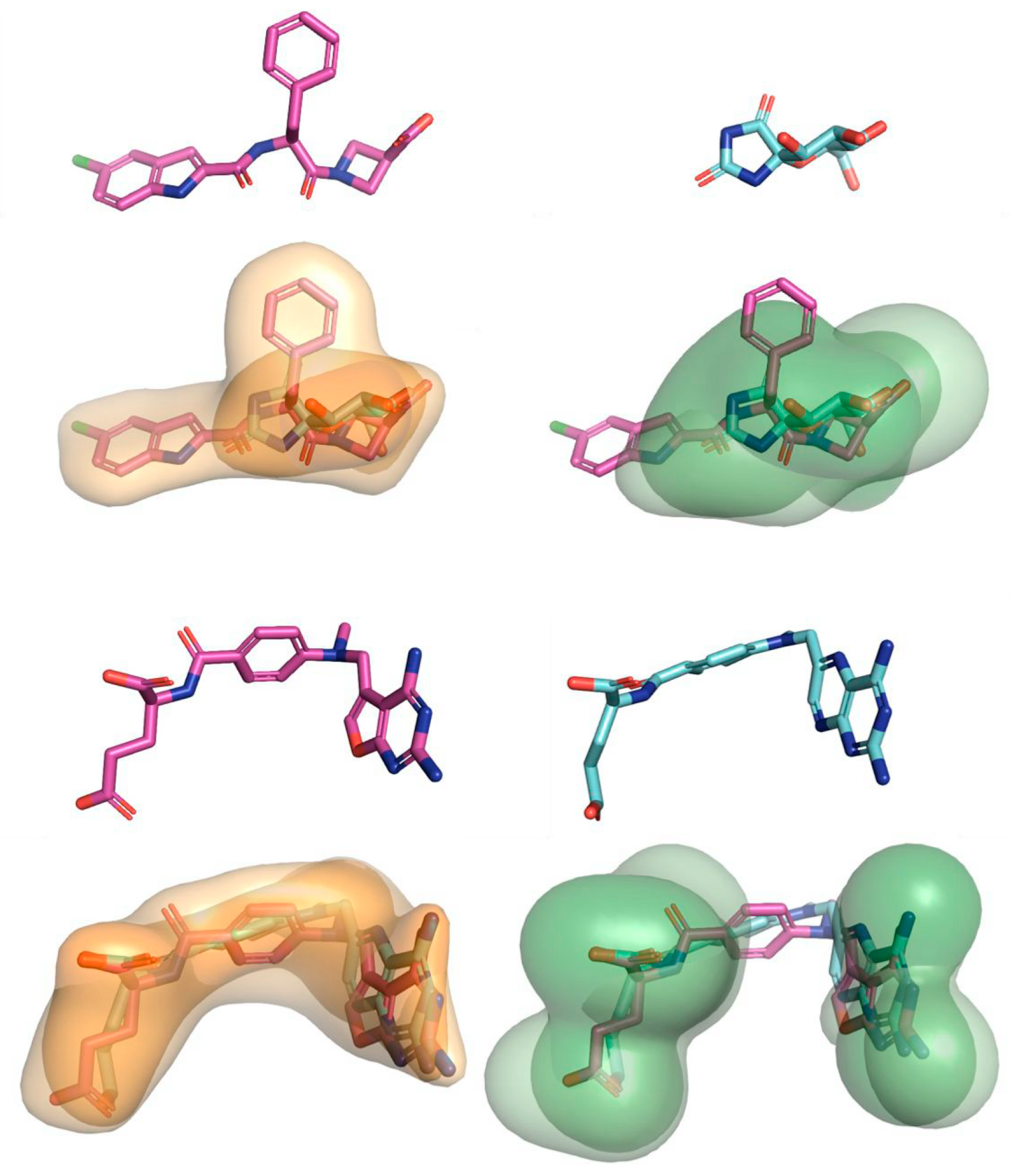
- Anighoro, A.; Bajorath, J. Three-dimensional similarity in molecular docking: Prioritizing ligand poses on the basis of experimental binding modes. J. Chem. Inf. Model. 2016, 56, 580–587. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J. A hybrid virtual screening protocol based on binding mode similarity. Methods Mol. Biol. 2018, 1824, 165–176. [Google Scholar] [PubMed]
- Jacquemard, C.; Drwal, M.N.; Desaphy, J.; Kellenberger, E. Binding mode information improves fragment docking. J. Cheminform. 2019, 11, 24. [Google Scholar] [CrossRef]
- Jacquemard, C.; Tran-Nguyen, V.-K.; Drwal, M.N.; Rognan, D.; Kellenberger, E. Local interaction density (LID), a fast and efficient tool to prioritize docking poses. Molecules 2019, 24, 2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez, J.; Deplano, A.; Herrero, A.; Gibert, E.; Herrero, E.; Luque, F.J. Assessing the performance of mixed strategies to combine lipophilic molecular similarity and docking in virtual screening. J. Chem. Inf. Model. 2020, 60, 4231–4245. [Google Scholar] [CrossRef]
- Ai, G.; Tian, C.; Deng, D.; Fida, G.; Chen, H.; Ding, L.; Ma, Y.; Gu, Y. A Combination of 2D similarity search, pharmacophore, and molecular docking techniques for the identification of vascular endothelial growth factor receptor-2 inhibitors. Anticancer. Drugs 2015, 26, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Staroń, J.; Kurczab, R.; Warszycki, D.; Satała, G.; Krawczyk, M.; Bugno, R.; Lenda, T.; Popik, P.; Hogendorf, A.S.; Hogendorf, A.; et al. Virtual screening-driven discovery of dual 5-HT6/5-HT2A receptor ligands with pro-cognitive properties. Eur. J. Med. Chem. 2020, 185, 111857. [Google Scholar] [CrossRef]
- Oum, Y.H.; Kell, S.A.; Yoon, Y.; Liang, Z.; Burger, P.; Shim, H. Discovery of novel aminopiperidinyl amide CXCR4 modulators through virtual screening and rational drug design. Eur. J. Med. Chem. 2020, 201, 112479. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.; Ahemad, N.; Chuah, L.H.; Naidu, R.; Htar, T.T. Sequential ligand- and structure-based virtual screening approach for the identification of potential g protein-coupled estrogen receptor-1 (GPER-1) modulators. RSC Adv. 2019, 9, 2525–2538. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Ren, J.; Ma, Y.; Liu, H.; Rong, Q.; Feng, Y.; Wang, Y.; Cheng, Y.; Ge, R.; Li, Z.; et al. Discovery of cyanopyridine scaffold as novel indoleamine-2,3-dioxygenase 1 (IDO1) inhibitors through virtual screening and preliminary hit optimisation. J. Enzyme Inhib. Med. Chem. 2019, 34, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Luo, G.; Qiao, L.; Jiang, L.; Li, G.; Zhang, Y. Virtual screening for potential allosteric inhibitors of cyclin-dependent kinase 2 from traditional chinese medicine. Molecules 2016, 2, 1259. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.W.; Wang, M.Y.; Wang, S.; Li, S.L.; Li, W.Q.; Meng, F.H. Identification of novel CDK2 inhibitors by a multistage virtual screening method based on SVM, pharmacophore and docking model. J. Enzyme Inhib. Med. Chem. 2020, 35, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Silakari, O. Ligand-based and e-pharmacophore modeling, 3D-QSAR and hierarchical virtual screening to identify dual inhibitors of spleen tyrosine kinase (Syk) and janus kinase 3 (JAK3). J. Biomol. Struct. Dyn. 2017, 35, 3043–3060. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Watanabe, E.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J. Comput. Aided Mol. Des. 2017, 31, 201–211. [Google Scholar] [CrossRef]
- Liu, K.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations: A cross-docking study. J. Chem. Inf. Model. 2017, 57, 2514–2522. [Google Scholar] [CrossRef]
- Mollica, L.; Theret, I.; Antoine, M.; Perron-Siera, F.; Charton, Y.; Fourquez, J.-M.; Wierzbicki, M.; Boutin, J.A.; Ferry, G.; Decherchi, S.; et al. Molecular dynamics simulations and kinetic measurements to estimate and predict protein-ligand residence times. J. Med. Chem. 2016, 59, 7167–7176. [Google Scholar] [CrossRef]
- Majewski, M.; Barril, X. Structural stability predicts the binding mode of protein-ligand complexes. J. Chem. Inf. Mod. 2020, 60, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- OpenEye Scientic Software. EON. 2.2.0.5. Santa Fe, NM, USA. Available online: https://www.eyesopen.com/ (accessed on 15 October 2020).
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Dawood, H.M.; Ibrahim, R.S.; Shawky, E.; Hammoda, H.M.; Metwally, A.M. Integrated in silico-in vitro strategy for screening of some traditional egyptian plants for human artomatase inhibitors. J. Ethnopharmacol. 2018, 224, 359–372. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3d database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Svensson, F.; Karlén, A.; Sköld, C. Virtual screening data fusion using both structure-and ligand-based methods. J. Chem. Inf. Model. 2012, 52, 225–232. [Google Scholar] [CrossRef]
- Willett, P. Enhancing the effectiveness of ligand-based virtual screening using data fusion. QSAR Comb. Sci. 2006, 25, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Willett, P. Combination of similarity rankings using data fusion. J. Chem. Inf. Model. 2013, 53, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, J.; Dilworth, D.; Reiser, U.; Neumüller, R.A.; Schleicher, M.; Petronczki, M.; Zeeb, M.; Mischerikow, N.; Allali-Hassani, A.; Szewczyk, M.M.; et al. Fragment-based discovery of a chemical probe for the PWWP1 domain of NSD3. Nat. Chem. Biol. 2019, 15, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Arany, A.; Bolgar, B.; Balogh, B.; Antal, P.; Matyus, P. Multi-aspect candidates for repositioning: Data fusion methods using heterogeneous information sources. Curr. Med. Chem. 2012, 20, 95–107. [Google Scholar] [CrossRef]
- Huang, G.; Li, J.; Wang, P.; Li, W. A review of computational drug repositioning approaches. Comb. Chem. High Throughput Screen. 2017, 20, 831–838. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Li, S.; Wang, Y.; Peng, J.; Luo, C.; Luo, X.; Zheng, M.; Chen, K.; Jiang, H. In silico target fishing: Addressing a “big data” Problem by ligand-based similarity rankings with data fusion. J. Cheminform. 2014, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bajusz, D.; Rácz, A.; Héberger, K. Comparison of data fusion methods as consensus scores for ensemble docking. Molecules 2019, 24, 2690. [Google Scholar] [CrossRef] [Green Version]
- Jaundoo, R.; Bohmann, J.; Gutierrez, G.; Klimas, N.; Broderick, G.; Craddock, T. Using a consensus docking approach to predict adverse drug reactions in combination drug therapies for gulf war illness. Int. J. Mol. Sci. 2018, 19, 3355. [Google Scholar] [CrossRef] [Green Version]
- Charifson, P.S.; Corkery, J.J.; Murcko, M.A.; Walters, W.P. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 1999, 42, 5100–5109. [Google Scholar] [CrossRef]
- Feher, M. Consensus scoring for protein-ligand interactions. Drug Discov. Today 2006, 11, 421–428. [Google Scholar] [CrossRef]
- Sun, H.; Pan, P.; Tian, S.; Xu, L.; Kong, X.; Li, Y.; Li, D.; Hou, T. Constructing and validating high-performance MIEC-SVM models in virtual screening for kinases: A better way for actives discovery. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.; Tian, S.; Pan, P.; Sun, H.; Li, D.; Li, Y.; Zhou, H.; Li, C.; Ming-Yuen Lee, S.; Hou, T. Discovery of novel ROCK1 inhibitors via integrated virtual screening strategy and bioassays. Sci. Rep. 2015, 5, 16749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.; Geppert, H.; Sisay, M.T.; Gütschow, M.; Bajorath, J. Integrating structure- and ligand-based virtual screening: Comparison of individual, parallel, and fused molecular docking and similarity search calculations on multiple targets. ChemMedChem 2008, 3, 1566–1571. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hahn, M. Extended-connectivity fingerpirints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Berry, M.; Fielding, B.C.; Gamieldien, J. Potential broad spectrum inhibitors of the coronavirus 3CLpro: A virtual screening and structure-based drug design study. Viruses 2015, 7, 6642–6660. [Google Scholar] [CrossRef] [Green Version]
- Vucicevic, J.; Srdic-Rajic, T.; Pieroni, M.; Laurila, J.M.M.; Perovic, V.; Tassini, S.; Azzali, E.; Costantino, G.; Glisic, S.; Agbaba, D.; et al. A combined ligand- and structure-based approach for the identification of rilmenidine-derived compounds which synergize the antitumor effects of doxorubicin. Bioorganic Med. Chem. 2016, 24, 3174–3183. [Google Scholar] [CrossRef]
- Jang, C.; Yadav, D.K.; Subedi, L.; Venkatesan, R.; Venkanna, A.; Afzal, S.; Lee, E.; Yoo, J.; Ji, E.; Kim, S.Y.; et al. Identification of novel acetylcholinesterase inhibitors designed by pharmacophore-based virtual screening, molecular docking and bioassay. Sci. Rep. 2018, 8, 14921. [Google Scholar] [CrossRef] [Green Version]
- Costa, G.; Rocca, R.; Corona, A.; Grandi, N.; Moraca, F.; Romeo, I.; Talarico, C.; Gagliardi, M.G.; Ambrosio, F.A.; Ortuso, F.; et al. Novel natural non-nucleoside inhibitors of HIV-1 reverse transcriptase identified by shape- and structure-based virtual screening techniques. Eur. J. Med. Chem. 2019, 161, 1–10. [Google Scholar] [CrossRef]
- Vedani, A.; Zbinden, P.; Snyder, J.P. Pseudo-receptor modeling: A new concept for the three-dimensional construction of receptor binding sites. J. Recept. Signal Transduct. 1993, 13, 163–177. [Google Scholar] [CrossRef]
- Andrews, P.R.; Quint, G.; Winkler, D.A.; Richardson, D.; Sadek, M.; Spurling, T.H. Morpheus: A conformation-activity relationships and receptor modeling package. J. Mol. Graph. 1989, 7, 138–145. [Google Scholar] [CrossRef]
- Pei, J.; Chen, H.; Liu, Z.; Han, X.; Wang, Q.; Shen, B.; Zhou, J.; Lai, L. Improving the quality of 3D-QSAR by using flexible-ligand receptor models. J. Chem. Inf. Model. 2005, 45, 1920–1933. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for ligands and proteins (FLAP): Theory and application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef]
- Jacob, L.; Vert, J.-P. Protein-ligand interaction prediction: An improved chemogenomics approach. Bioinformatics 2008, 24, 2149–2156. [Google Scholar] [CrossRef] [Green Version]
- Salam, N.K.; Nuti, R.; Sherman, W. Novel method for generating structure-based pharmacophores using energetic analysis. J. Chem. Inf. Model. 2009, 49, 2356–2368. [Google Scholar] [CrossRef]
- Sato, T.; Honma, T.; Yokoyama, S. Combining machine learning and pharmacophore-based interaction fingerprint for in silico screening. J. Chem. Inf. Model. 2010, 50, 170–185. [Google Scholar] [CrossRef]
- Koes, D.R.; Camacho, C.J. ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Res. 2012, 40, W409–W414. [Google Scholar] [CrossRef]
- Tran-Nguyen, V.K.; Da Silva, F.; Bret, G.; Rognan, D. All in one: Cavity detection, druggability estimate, cavity-based pharmacophore perception, and virtual screening. J. Chem. Inf. Model. 2019, 59, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Chuaqui, C.; Singh, J. Structural interaction fingerprint (SIFt): A novel method for analyzing three-dimensional protein-ligand binding interactions. J. Med. Chem. 2004, 47, 337–344. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, 443–447. [Google Scholar] [CrossRef]
- Hajiebrahimi, A.; Ghasemi, Y.; Sakhteman, A. FLIP: An assisting software in structure based drug design using fingerprint of protein-ligand interaction profiles. J. Mol. Graph. Model. 2017, 78, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Jasper, J.B.; Humbeck, L.; Brinkjost, T.; Koch, O. A novel interaction fingerprint derived from per atom score contributions: Exhaustive evaluation of interaction fingerprint performance in docking based virtual screening. J. Cheminform. 2018, 10, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, F.; Desaphy, J.; Rognan, D. IChem: A versatile toolkit for detecting, comparing, and predicting protein-ligand interactions. ChemMedChem 2018, 13, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Desaphy, J.; Raimbaud, E.; Ducrot, P.; Rognan, D. Encoding protein-ligand interaction patterns in fingerprints and graphs. J. Chem. Inf. Model. 2013, 53, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Salentin, S.; Adasme, M.F.; Heinrich, J.C.; Haupt, V.J.; Daminelli, S.; Zhang, Y.; Schroeder, M. From malaria to cancer: Computational drug repositioning of amodiaquine using PLIP interaction patterns. Sci. Rep. 2017, 7, 11401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Surflex-Dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J. Comput. Aided Mol. Des. 2007, 21, 281–306. [Google Scholar] [CrossRef] [Green Version]
- Symyx Software. MACCS Structural Keys; Symyx Technologies: San Ramon, CA, USA, 2002. [Google Scholar]
- Jubb, H.C.; Higueruelo, A.P.; Ochoa-Montaño, B.; Pitt, W.R.; Ascher, D.B.; Blundell, T.L. Arpeggio: A web server for calculating and visualising interatomic interactions in protein structures. J. Mol. Biol. 2017, 429, 365–371. [Google Scholar] [CrossRef]
- Marcou, G.; Rognan, D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J. Chem. Inf. Model. 2007, 47, 195–207. [Google Scholar] [CrossRef]
- Pérez-Nueno, V.I.; Rabal, O.; Borrell, J.I.; Teixidó, J. APIF: A new interaction fingerprint based on atom pairs and its application to virtual screening. J. Chem. Inf. Model. 2009, 49, 1245–1260. [Google Scholar] [CrossRef]
- Lenselink, E.B.; Jespers, W.; Van Vlijmen, H.W.T.; IJzerman, A.P.; Van Westen, G.J.P. Interacting with GPCRs: Using interaction fingerprints for virtual screening. J. Chem. Inf. Model. 2016, 56, 2053–2060. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Huang, S.; Mei, H.; Kevin, M.; Shi, T.; Chen, L. Protein–ligand interaction fingerprints for accurate prediction of dissociation rates of P38 MAPK type II inhibitors. Integr. Biol. 2019, 11, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Bourne, P.E. Revealing acquired resistance mechanisms of kinase-targeted drugs using an on-the-fly, function-site interaction fingerprint approach. J. Chem. Theory Comput. 2020, 16, 3152–3161. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, K.Y.J. Application of shape similarity in pose selection and virtual screening in CSARdock2014 exercise. J. Chem. Inf. Model. 2016, 56, 965–973. [Google Scholar] [CrossRef]
- Prathipati, P.; Mizuguchi, K. Integration of ligand and structure based approaches for CSAR-2014. J. Chem. Inf. Model. 2016, 56, 974–987. [Google Scholar] [CrossRef]
- Kumar, A.; Zhang, K.Y.J. A pose prediction approach based on ligand 3D shape similarity. J. Comput. Aided Mol. Des. 2016, 30, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, K.Y.J. Improving ligand 3D shape similarity-based pose prediction with a continuum solvent model. J. Comput. Aided Mol. Des. 2019, 33, 1045–1055. [Google Scholar] [CrossRef]
- Kumar, A.; Zhang, K.Y.J. Prospective evaluation of shape similarity based pose prediction method in D3R grand challenge 2015. J. Comput. Aided Mol. Des. 2016, 30, 685–693. [Google Scholar] [CrossRef]
- Lee, H.S.; Choi, J.; Kufareva, I.; Abagyan, R.; Filikov, A.; Yang, Y.; Yoon, S. Optimization of high throughput virtual screening by combining shape-matching and docking methods. J. Chem. Inf. Model. 2008, 48, 489–497. [Google Scholar] [CrossRef]
- Kelley, B.P.; Brown, S.P.; Warren, G.L.; Muchmore, S.W. POSIT: Flexible shape-guided docking for pose prediction. J. Chem. Inf. Model. 2015, 55, 1771–1780. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J. Binding mode similarity measures for ranking of docking poses: A case study on the adenosine A2Areceptor. J. Comput. Aided Mol. Des. 2016, 30, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Anighoro, A.; Bajorath, J. Compound ranking based on fuzzy three-dimensional similarity improves the performance of docking into homology models of g-protein-coupled receptors. ACS Omega 2017, 2, 2583–2592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marialke, J.; Tietze, S.; Apostolakis, J. Similarity based docking. J. Chem. Inf. Model. 2008, 48, 186–196. [Google Scholar] [CrossRef]
- Gathiaka, S.; Liu, S.; Chiu, M.; Yang, H.; Stuckey, J.A.; Kang, Y.N.; Delproposto, J.; Kubish, G.; Dunbar, J.B.; Carlson, H.A.; et al. D3R grand challenge 2015: Evaluation of protein–ligand pose and affinity predictions. J. Comput. Aided Mol. Des. 2016, 30, 651–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaieb, Z.; Liu, S.; Gathiaka, S.; Chiu, M.; Yang, H.; Shao, C.; Feher, V.A.; Walters, W.P.; Kuhn, B.; Rudolph, M.G.; et al. D3R grand challenge 2: Blind prediction of protein–ligand poses, affinity rankings, and relative binding free energies. J. Comput. Aided Mol. Des. 2018, 32, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Gaieb, Z.; Parks, C.D.; Chiu, M.; Yang, H.; Shao, C.; Walters, W.P.; Lambert, M.H.; Nevins, N.; Bembenek, S.D.; Ameriks, M.K.; et al. D3R grand challenge 3: Blind prediction of protein-ligand poses and affinity rankings. J. Comput. Aided Mol. Des. 2019, 33, 1–18. [Google Scholar] [CrossRef]
- Kumar, A.; Zhang, K.Y.J. Shape similarity guided pose prediction: Lessons from D3R grand challenge 3. J. Comput. Aided Mol. Des. 2019, 33, 47–59. [Google Scholar] [CrossRef]
- Kumar, A.; Zhang, K.Y.J. A cross docking pipeline for improving pose prediction and virtual screening performance. J. Comput. Aided Mol. Des. 2018, 32, 163–173. [Google Scholar] [CrossRef]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef]
- Varela-Rial, A.; Majewski, M.; Cuzzolin, A.; Martinez-Rosell, G.; De Fabritiis, G. SkeleDock: A web application for scaffold docking in PlayMolecule. J. Chem. Inf. Model. 2020, 60, 2673–2677. [Google Scholar] [CrossRef]
- Marialke, J.; Körner, R.; Tietze, S.; Apostolakis, J. Graph-based molecular alignment (GMA). J. Chem. Inf. Model. 2007, 47, 591–601. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Molecular Operating Environment 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2020.
- Ginex, T.; Vázquez, J.; Gibert, E.; Herrero, E.; Luque, F.J. Lipophilicity in drug design: An overview of lipophilicity descriptors in 3D-QSAR. Fut. Med. 2019, 11, 1177–1193. [Google Scholar] [CrossRef] [PubMed]
- PharmScreen. Pharmacelera, Barcelona. 2019. Available online: www.pharmacelera.com (accessed on 15 October 2020).
- Ruiz-Carmona, S.; Alvarez-García, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A Dast, Versatile and open source porgram for docking ligands to proteins and nucleic acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.; Willet, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Kogej, T.; Engkvist, O. Cheminformatics in drug discovery, an industrial perspective. Mol. Inform. 2018, 37, 1800041. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Patrícia Bento, A.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrí an-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2016, 45, 945–954. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, 623–633. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the drugbank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Hoffmann, T.; Gastreich, M. The next level in chemical space navigation: Going far beyond enumerable compound libraries. Drug Discov. Today 2019, 24, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Van Hilten, N.; Chevillard, F.; Kolb, P. Virtual compound libraries in computer-assisted drug discovery. J. Chem. Inf. Model. 2019, 59, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Miyao, T.; Arakawa, M.; Funatsu, K. Exhaustive structure generation for inverse-QSPR/QSAR. Mol. Inform. 2010, 29, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Hartenfellar, M.; Proschak, E.; Schüller, G. DOGS: Reaction-driven de novo design of bioactive compounds. PLoS Comput. Biol. 2012, 8, 1–12. [Google Scholar] [CrossRef]
- Pottel, J.; Moitessier, N. Customizable generation of synthetically accessible, local chemical subspaces. J. Chem. Inf. Model. 2017, 57, 454–467. [Google Scholar] [CrossRef]
- Gao, W.; Coley, C.W. The synthesizability of molecules proposed by generative models. J. Chem. Inf. Model. 2020. [Google Scholar] [CrossRef] [Green Version]
- Mishima, K.; Kaneko, H.; Funatsu, K. Development of a new de novo design algorithm for exploring chemical space. Mol. Inform. 2014, 33, 779–789. [Google Scholar] [CrossRef]
- Podlewska, S.; Czarnecki, W.M.; Kafel, R.; Bojarski, A.J. Creating the new from the old: Combinatorial libraries generation with machine-learning-based compound structure optimization. J. Chem. Inf. Model. 2017, 57, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Nguyen, D.D.; Tu, M.; Wei, G.-W. Generative network complex for the automated generation of drug-like molecules. J. Chem. Inf. Model. 2020. [Google Scholar] [CrossRef]
- Amabilino, S.; Pogány, P.; Pickett, S.D.; Green, D.V.S. Guidelines for recurrent neural network transfer learning-based molecular generation of focused libraries. J. Chem. Inf. Model. 2020. [Google Scholar] [CrossRef]
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular de-novo design through deep reinforcement learning. J. Cheminform. 2017, 9, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domenico, A.; Nicola, G.; Daniela, T.; Fulvio, C.; Nicola, A.; Orazio, N. De novo drug design of targeted chemical libraries based on artificial intelligence and pair-based multiobjective optimization. J. Chem. Inf. Model. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J. PPI inhibitor and stabilizer development in human diseases. Drug Discov. Today Technol. 2017, 24, 3–9. [Google Scholar] [CrossRef]
- Sijbesma, E.; Hallenbeck, K.K.; Leysen, S.; De Vink, P.J.; Skóra, L.; Jahnke, W.; Brunsveld, L.; Arkin, M.R.; Ottmann, C. Site-directed fragment-based screening for the discovery of protein-protein interaction stabilizers. J. Am. Chem. Soc. 2019, 141, 3524–3531. [Google Scholar] [CrossRef]
- Stevers, L.M.; Sijbesma, E.; Botta, M.; Mackintosh, C.; Obsil, T.; Landrieu, I.; Cau, Y.; Wilson, A.J.; Karawajczyk, A.; Eickhoff, J.; et al. Modulators of 14-3-3 protein-protein interactions. J. Med. Chem. 2018, 61, 3755–3778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petta, I.; Lievens, S.; Libert, C.; Tavernier, J.; De Bosscher, K. Modulation of protein-protein interactions for the development of novel therapeutics. Mol. Ther. 2016, 24, 707–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, M.; Lee, G.M.; Sijbesma, E.; Ottman, C.; Arkin, M.R. Modulating protein-protein interaction networks in protein homeostasis. Curr. Opin. Chem. Biol. 2019, 50, 55–65. [Google Scholar] [CrossRef]
- Reynès, C.; Host, H.; Camproux, A.-C.; Laconde, G.; Leroux, F.; Mazars, A.; Deprez, B.; Fahraeus, R.; Villoutreix, B.O.; Sperandio, O. Designing focused chemical libraries enriched in protein-protein interaction inhibitors using machine-learning methods. PLoS Comput. Biol. 2010, 6, e1000695. [Google Scholar] [CrossRef]
- Hamon, V.; Brunel, J.-M.; Combes, S.; Basse, M.J.; Roche, P.; Morelli, X. 2P2IChem: Focused chemical libraries dedicated to orthosteric modulation of protein-protein interactions. MedChemComm 2013, 4, 797–809. [Google Scholar] [CrossRef]
- Bosc, N.; Muller, C.; Hoffer, L.; Lagorce, D.; Bourg, S.; Derviaux, C.; Gourdel, M.-E.; Rain, J.-C.; Miller, T.W.; Villoutreix, B.O.; et al. Fr-PPIChem: An academic compound library dedicated to protein-protein interactions. ACS Chem. Biol. 2020, 15, 1566–1574. [Google Scholar] [CrossRef]
- Zhang, X.; Betzi, S.; Morelli, X.; Roche, P. Focused chemical libraries—Design and enrichment: An example of protein-protein interaction chemical space. Future Med. Chem. 2014, 6, 1291–1307. [Google Scholar] [CrossRef]
- Singh, N.; Chaput, L.; Villoutreix, B.O. Fast rescoring protocols to improve the performance of structure-based virtual screening performed on protein-protein interfaces. J. Chem. Inf. Model. 2020, 60, 3910–3934. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Monteagudo, M.; Schürer, S.; Tejera, E.; Pérez-Castillo, Y.; Medina-Franco, J.L.; Sánchez-Rodríguez, A.; Borges, F. Systemic QSAR and phenotypic virtual screening: Chasing butterflies in drug discovery. Drug Discov. Today 2017, 22, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.Y.; Simeonov, A.; Jadhav, A.; Babaoglu, K.; Inglese, J.; Shoichet, B.K.; Austin, C.P. A High-throughput screen for aggregation-based inhibition in a large compound library. J. Med. Chem. 2007, 50, 2385–2390. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.Y.; Shoichet, B.K. A Detergent-based assay for the detection of promiscous inhibitors. Nat. Protoc. 2006, 1, 550–553. [Google Scholar] [CrossRef]
- Duan, D.; Torosyan, H.; Elnatan, D.; McLaughlin, C.K.; Logie, J.; Shoichet, M.S.; Agard, D.A.; Shoichet, B.K. Internal structure and preferential protein binding of colloidal aggregates. ACS Chem. Biol. 2017, 12, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Owen, S.C.; Doak, A.K.; Wassam, P.; Schoichet, M.S.; Schoichet, B.K. Colloidal aggregation affects the efficacy of anticancer drugs in cell culture. ACS Chem. Biol. 2012, 7, 1249–1435. [Google Scholar] [CrossRef]
- Dlim, M.M.; Shahout, F.S.; Khabir, M.K.; Labonté, P.P.; LaPlante, S.R. Revealing drug self-associations into nano-entities. ACS Omega 2019, 4, 8919–8925. [Google Scholar] [CrossRef]
- Liu, Y.; Beresini, M.H.; Johnson, A.; Mintzer, R.; Shah, K.; Clark, K.; Schmidt, S.; Lewis, C.; Liimatta, M.; Elliott, L.O.; et al. Case studies of minimizing nonspecific inhibitors in HTS campaigns that use assay-ready plates. J. Biomol. Screen. 2012, 17, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Ghattas, M.A.; Al Rawashdeh, S.; Atatreh, N.; Bryce, R.A. How do small molecule aggregates inhibit enzyme activity? A molecular dynamics study. J. Chem. Inf. Model. 2020, 60, 3901–3909. [Google Scholar] [CrossRef]
- Coan, K.E.D.; Schoichet, B.K. Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc. 2008, 130, 9606–9612. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Algorithm | Description | Case Studies |
|---|---|---|
| ADDITION algorithms | Adds together the ranks from the different VS methods rank lists. Standard statistical measures, weighted or not, are used (i.e., sum, average, and median or max. value) to combine rank positions. | [66,67,103,116] |
| PARETO ranking | Ranks a compound based on how many other compounds are better in all screening methods. Ties could be broken using the sum rank, as example. | [103] |
| PARALLEL selection | Compounds are alternatively selected among the top-ranked compounds obtained from each screening method until the desired number of compounds is reached. | [81,103] |
| Model | Methods | Description |
|---|---|---|
| Interaction fingerprint-based | SIFt [132] | Fingerprint encoding seven predefined types of target–ligand interactions. |
| PLIP [133] | A web service for the detection and visualization of seven protein–ligand interaction types considering a 3D space. | |
| FLIP [134] | For each residue, 7 different interactions are represented in 10 bits. | |
| PADIF [135] | Fingerprints with the inclusion of information relative to the strength of interactions and unfavorable ones. | |
| Pharmacophore-based | LigandScout [125] | Pharmacophores derived from six types of nonbonded protein-ligand interactions and volume constraints. |
| FLAP [126] | Four-point pharmacophore fingerprints with a shape component. | |
| IChem [136] | Converts the protein–ligand interaction pattern in fingerprints and graphs. | |
| TIFP [137] | Encodes a string of unique triplets (two interacting atoms and an interaction pseudo-atom). |
| Database | Type | No. Cpds |
|---|---|---|
| AstraZeneca with Enamine BBs | Proprietary | 1017 |
| Boehr.-Ing. BICLAIM | Proprietary | 5 × 1011 |
| CH/PMUNK | Public | >95 × 106 |
| eMolecules Plus | Commercial | 5.9 × 108 |
| Enamine Real | Commercial | >300 × 106 |
| EVOspace | Proprietary | 1.6 × 1016 |
| GDB-17 | Public | ~166 × 109 |
| Lilly LPC | Proprietary | 2 × 1011 |
| MASSIV | Proprietary | 1020 |
| SAVI | Public | ~283 × 106 |
| PGVL | Proprietary | 3 × 1012 |
| PubChem | Public | 9.6 × 106 |
| SCUBIDOO | Public | ~21 × 106 |
| Sigma Aldrich | Commercial | 1.4 × 107 |
| ZINC15 | Commercial | 2 × 106 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez, J.; López, M.; Gibert, E.; Herrero, E.; Luque, F.J. Merging Ligand-Based and Structure-Based Methods in Drug Discovery: An Overview of Combined Virtual Screening Approaches. Molecules 2020, 25, 4723. https://doi.org/10.3390/molecules25204723
Vázquez J, López M, Gibert E, Herrero E, Luque FJ. Merging Ligand-Based and Structure-Based Methods in Drug Discovery: An Overview of Combined Virtual Screening Approaches. Molecules. 2020; 25(20):4723. https://doi.org/10.3390/molecules25204723
Chicago/Turabian StyleVázquez, Javier, Manel López, Enric Gibert, Enric Herrero, and F. Javier Luque. 2020. "Merging Ligand-Based and Structure-Based Methods in Drug Discovery: An Overview of Combined Virtual Screening Approaches" Molecules 25, no. 20: 4723. https://doi.org/10.3390/molecules25204723
APA StyleVázquez, J., López, M., Gibert, E., Herrero, E., & Luque, F. J. (2020). Merging Ligand-Based and Structure-Based Methods in Drug Discovery: An Overview of Combined Virtual Screening Approaches. Molecules, 25(20), 4723. https://doi.org/10.3390/molecules25204723