4.2. Synthesis
2′,3′,5′-Tri-O-acetyl-8-bromoadenosine (6). The title compound was obtained by peracetylation of commercially available 8-bromoadenosine (90% yield).
6-Chloro-9-(2′,3′-O-isopropylidene-ß-D-ribofuranosyl)purine (10). The title compound was obtained by reaction of 6-chloropurine riboside with 2,2-dimethoxypropane in acetone in the presence of toluene-4-sulfonic acid for 3 h at room temperature (91% yield) [
22].
N6-(2-tert-Butyloxycarbonyl)aminoethyl-2’,3’-O-isopropylidene-adenosine (11a). To a stirred solution of 6-chloro-9-(2′,3′-
O-isopropylidene-
ß-
d-ribofuranosyl)purine (
10) (0.98 g, 3.0 mmol) in anhydrous DME (15 mL) was added NEt
3 (1.25 mL, 9.0 mmol), followed by a solution of
N-Boc-ethylenediamine (prepared according to reported procedures) [
23] (1.44 g, 9.0 mmol) in anhydrous DME (15 mL). The stirred mixture was heated at 80 °C overnight under an argon atmosphere, then concentrated under reduced pressure. The residue was dissolved in dichloromethane (80 mL), washed in turn with water (80 mL) and brine (80 mL). The organic layer was dried over Na
2SO
4 and concentrated under reduced pressure. The crude product was purified by flash column chromatography (50 g SiO
2, 0 to 3% methanol in dichloromethane) to afford compound
11a (0.90 g, 66%) as a white foam.
1H NMR (400 MHz, CDCl
3):
δ 1.40 (s, 3H, CH
3 isop), 1.44 (s, 9H, CH
3 Boc), 1.66 (s, 3H, CH
3 isop), 3.44 -3.48 (m, 2H,
CH2NHBoc), 3.76-3.84 (m, 3H,
CH2NH-6 and H-5’), 3.99 (dd,
J = 1.4 Hz,
J = 12.8 Hz, 1H, H-5”), 4.53-4.56 (m, 1H, H-4’), 5.09 (br s, 1H, NHBoc), 5.13 (dd,
J = 5.8 Hz,
J = 1.0 Hz, 1H, H-3’), 5.21 (pt,
J = 5.5 Hz, 1H, H-2’), 5.87 (d,
J = 4.9 Hz, 1H, H-1’), 6.40-6.90 (br s, 2H, NH-6 and OH-5’), 7.84 (s, 1H, H-8), 8.33 (s, 1H, H-2);
13C NMR (100 MHz, CDCl
3):
δ 25.25 (CH
3 isop), 27.66 (CH
3 isop), 28.34 (3 CH
3 Boc), 40.74 (
CH2NH-6 and
CH2NHBoc), 63.42 (C-5’), 79.59 (Cq Boc), 81.71 (C-3’), 83.09 (C-2’), 86.15 (C-4’), 94.32 (C-1’), 113.98 (Cq isop), 121.42 (C-5), 139.76 (C-8), 147.61 (C-4), 152.91 (C-2), 155.35 (C-6), 156.30 (CO Boc); HRMS (ESI-TOF):
m/z calcd for [C
20H
30N
6O
6+H]
+ 451.2305, found 451.2293.
N6-(2-tert-Butyloxycarbonyl)aminobutyl-2’,3’-O-isopropylidene-adenosine (11b). Compound
11b was prepared from
10 (0.98 g, 3.0 mmol) and
N-Boc-1,4-butanediamine (prepared according to reported procedures) [
23] (1.69 g, 9.0 mmol) following the same procedure as for
11a. Purification by flash column chromatography (50 g SiO
2, 0 to 3% methanol in dichloromethane) yielded compound
11b (1.31 g, 91%) as a white foam.
1H NMR (400 MHz, CDCl
3):
δ 1.39 (s, 3H, CH
3 isop), 1.45 (s, 9H, CH
3 Boc), 1.61 (quint,
J = 7.0 Hz, 2H, CH
2), 1.66 (s, 3H, CH
3 isop), 1.74 (quint,
J = 7.0 Hz, 2H, CH
2), 3.19 (br q, 2H,
CH2NHBoc), 3.68 (br s, 2H,
CH2NH-6), 3.80 (br d,
J = 12.8 Hz, 1H, H-5’), 3.99 (dd,
J = 1.3 Hz,
J = 12.8 Hz, 1H, H-5”), 4.53-4.56 (m, 1H, H-4’), 4.78 (br s, 1H, NHBoc), 5.13 (dd,
J = 1.0 Hz,
J = 5.9 Hz, 1H, H-3’), 5.22 (pt,
J = 5.8 Hz, 1H, H-2’), 5.86 (d,
J = 4.8 Hz, 1H, H-1’), 6.04 (br s, 1H, OH-5’), 6.67 (br s, 1H, NH-6), 7.79 (s, 1H, H-8), 8.33 (s, 1H, H-2);
13C NMR (100 MHz, CDCl
3):
δ 25.34 (CH
3 isop), 26.97 (CH
2), 27.24 (CH
2), 27.65 (CH
3 isop), 28.42 (CH
3 Boc), 40.16 (
CH2NH-6 and
CH2NHBoc), 63.42 (C-5’), 79.15 (C
q Boc), 81.72 (C-3’), 83.06 (C-2’), 86.14 (C-4’), 94.35 (C-1’), 113.94 (C
q isop), 121.25 (C-5), 139.52 (C-8), 147.31 (C-4), 152.69 (C-2), 155.25 (C-6), 156.01 (CO Boc); HRMS (ESI-TOF):
m/z calcd for [C
22H
34N
6O
6+H]
+ 479.2618, found 479.2614.
N6-(2-tert-Butyloxycarbonyl)aminoethyl-2’,3’-O-isopropylidene-5’-O-propargyl-adenosine (8a). To a stirred suspension of sodium hydride (0.10 g 60% in oil, 2.5 mmol) in anhydrous THF (12 mL) was added at 0 °C a solution of 11a (0.56 g, 1.2 mmol) in THF (12 mL). The reaction mixture was stirred at 0 °C for 1 h, then propargyl bromide (80% in toluene, 0.13 mL, 1.2 mmol) was added dropwise. After 24 h at 4 °C, the reaction was quenched by adding glacial acetic acid (0.14 mL) and stirring for 1 h at 4 °C. After removal of the volatiles under reduced pressure, the crude residue was dissolved ethyl acetate (50 mL) and washed twice with water (60 mL). The organic layer was dried, concentrated under reduced pressure and the residue purified by flash column chromatography (30 g SiO2, 0 to 3% methanol in dichloromethane) affording compound 8a (0.35 g, 60%) as a white foam. 1H NMR (400 MHz, CDCl3): δ 1.41 (s, 3H, CH3 isop), 1.43 (s, 9H, CH3 Boc), 1.64 (s, 3H, CH3 isop), 2.44 (t, J = 2.3 Hz, 1H, HC≡), 3.44 (br q, J = 5.6 Hz, 2H, CH2NHBoc), 3.73 (dd, J = 4.8 Hz, J = 10.3 Hz, 1H, H-5’), 3.78 (dd, J = 3.9 Hz, J = 10.3 Hz, 1H, H-5”), 3.81-3.85 (m, 2H, CH2NH-6), 4.14 (d, J = 2.3 Hz, 2H, CH2C≡), 4.49-4.53 (m, 1H, H-4’), 5.03 (dd, J = 2.7 Hz, J = 6.2 Hz, 1H, H-3’), 5.17 (br s, 1H, NHBoc), 5.34 (dd, J = 2.3 Hz, J = 6.2 Hz, 1H, H-2’), 6.19 (d, J = 2.4 Hz, 1H, H-1’), 6.33 (br s, 1H, NH-6), 8.01 (s, 1H, H-8), 8.39 (s, 1H, H-2); 13C NMR (100 MHz, CDCl3): δ 25.38 (CH3 isop), 27.18 (CH3 isop), 28.35 (3 CH3 Boc), 40.90 (CH2NH-6 and CH2NHBoc), 58.57 (C≡C-CH2), 69.85 (C-5’), 75.24 (C≡C-CH2), 78.80 (C≡C-CH2), 79.44 (Cq Boc), 81.88 (C-3’), 84.76 (C-2’), 85.74 (C-4’), 91.20 (C-1’), 114.23 (Cq isop), 120.16 (C-5), 138.93 (C-8), 148.87 (C-4), 152.91 (C-2), 154.88 (C-6), 156.27 (CO Boc); HRMS (ESI-TOF): m/z calcd for [C23H32N6O6+H]+ 489.2462, found 489.2463.
N6-(2-tert-Butyloxycarbonyl)aminobutyl-2’,3’-O-isopropylidene-5’-O-propargyl-adenosine (8b). Compound 8b was prepared from 11b (0.95 g, 2.0 mmol) following the same procedure as for 8a. Purification by flash column chromatography (70 g SiO2, 0 to 2% methanol in dichloromethane) afforded compound 8b (0.70 g, 68%). 1H NMR (400 MHz, CDCl3): δ 1.36 (s, 3H, CH3 isop), 1.41 (s, 9H, CH3 Boc), 1.51-1.63 (m, 2H, CH2), 1.59 (s, 3H, CH3 isop), 1.69 (quint, J = 7.0 Hz, 2H, CH2), 2.42 (t, J = 2.3 Hz, 1H, HC≡), 3.10-3.19 (m, 2H, CH2NHBoc), 3.60-3.66 (m, 2H, CH2NH-6), 3.69 (dd, J = 4.9 Hz, J = 10.3 Hz, 1H, H-5’), 3.74 (dd, J = 4.0 Hz, J = 10.3 Hz, 1H, H-5”), 4.10 (d, J = 2.3 Hz, 2H, CH2C≡), 4.44-4.49 (m, 1H, H-4’), 4.99 (dd, J = 2.6 Hz, J = 6.1 Hz, 1H, H-3’), 5.32 (dd, J = 2.3 Hz, J = 6.3 Hz, 1H, H-2’), 6.05 (br s, 1H, NH-6), 6.15 (d, J = 2.3 Hz, 1H, H-1’), 7.94 (s, 1H, H-8), 8.34 (s, 1H, H-2); 13C NMR (100 MHz, CDCl3): δ 25.34 (CH3 isop), 27.08 (CH3 isop), 27.14 and 27.19 (2 CH2), 28.40 (3 CH3 Boc), 40.12 (CH2NH-6 and CH2NHBoc), 58.53 (C≡C-CH2), 69.82 (C-5’), 75.30 (C≡C-CH2), 78.83 (C≡C-CH2), 78.99 (Cq Boc), 81.85 (C-3’), 84.68 (C-2’), 85.68 (C-4’), 91.06 (C-1’), 114.17 (Cq isop), 120.07 (C-5), 138.66 (C-8), 148.68 (C-4), 153.23 (C-2), 154.94 (C-6), 156.04 (CO Boc); HRMS (ESI-TOF): m/z calcd for [C25H36N6O6+H]+ 517.2775, found 517.2780.
2’,3’,5’-Tri-O-acetyl-8-[3-(N6-(2-tert-butyloxycarbonyl)aminoethyl-2’,3’-O-isopropylidene-5’-O-adenosinyl)propargyl]adenosine (12a). To an argon-degassed solution (three times) of 8a (0.20 g, 0.42 mmol) and bromide 6 (0.31 g, 0.63 mmol) in anhydrous THF (8 mL) in the presence of NEt3 (0.18 mL, 1.26 mmol) were added CuI (8 mg, 0.042 mmol) and Pd(PPh3)4 (24 mg, 0.021 mmol). The stirred mixture was degassed 3 times, then heated at 60 °C under argon atmosphere for 5 h. The volatiles were removed under reduced pressure and the residue purified by flash column chromatography (20 g SiO2, 0 to 4% methanol in dichloromethane) affording compound 12a (0.15 g, 40%) and the homocoupling product (63 mg, 8%). 1HNMR (400 MHz, CDCl3): δ 1.41 (s, 3H, CH3 isop), 1.43 (s, 9H, CH3 Boc), 1.64 (s, 3H, CH3 isop), 2.05 (s, 3H, COCH3), 2.10 (s, 3H, COCH3), 2.14 (s, 3H, COCH3), 3.39-3.47 (m, 2H, CH2NHBoc) 3.77 (br, 2H, CH2NH-6), 3.84 (dd, J = 5.1 Hz, J = 10.2 Hz, 1H, H-5’B), 3.92 (dd, J = 4.0 Hz, J = 10.2 Hz, 1H, H-5”B), 4.32 (dd, J = 6.0 Hz, J = 11.8 Hz, 1H, H-5’A), 4.41 (td, J = 6.0 Hz, J = 11.8 Hz, 1H, H-4’A), 4.47 (br, 2H, CH2C≡), 4.50-4.55 (m, 2H, H-4’B and H-5”A), 5.07 (dd, J = 3.0 Hz, J = 6.2 Hz, 1H, H-3’B), 5.23 (br s, 1H, NHBoc), 5.35-5.41 (br s, 1H, H-2’B), 5.96 (t, J = 6.1 Hz, 1H, H-3’A), 6.19 (d, J = 2.2 Hz, 1H, H-1’B), 6.22 (d, J = 4.2 Hz, 1H, H-1’A), 6.24 (br s, 2H, NH2), 6.30 (dd, J = 4.2 Hz, J = 6.1 Hz, 1H, H-2’A), 6.46 (br s, 1H, NH-6B), 8.02 (s, 1H, H-8B), 8.37 (s, 1H, H-2A), 8.42 (s, 1H, H-2B); 13CNMR (100 MHz, CDCl3): δ 20.43 (COCH3), 20.49 (COCH3), 20.64 (COCH3), 25.36 (CH3 isop), 27.16 (CH3 isop), 28.36 (3 CH3 Boc), 40.71 (CH2NHBoc and CH2NH-6), 59.04 (C≡C-CH2), 63.05 (C-5’A), 70.49 (C-5’B), 70.54 (C-3’A), 72.52 (C-2’A), 76.69 (C≡C-CH2), 79.49 (Cq Boc), 79.86 (C-4’A), 81.80 (C-3’B), 84.70 (C-2’B), 85.74 (C-4’B), 87.37 (C-1’A), 91.12 (C-1’B), 91.80 (C≡C-CH2), 114.36 (Cq isop), 119.78 (C-5A), 120.20 (C-5B), 133.26 (C-8A), 138.88 (C-8B); 148.90 (C-4B); 149.50 (C-4A), 153.23 (C-2B), 154.26 (C-2A), 155.00 (C-6B), 155.52 (C-6A), 156.31 (CO Boc), 169.42 (CO), 169.50 (CO), 170.58 (CO); HRMS (ESI-TOF): m/z calcd for [C39H49N11O13+H]+ 880.3589, found 880.3582.
2’,3’,5’-Tri-O-acetyl-8-[3-(N6-(4-tert-butyloxycarbonyl)aminobutyl-2’,3’-O-isopropylidene-5’-O-adenosinyl)propargyl]adenosine (12b). Compound 12b was prepared from 8b (206 mg, 0.4 mmol) and bromide 6 (184 mg, 0.26 mmol) following the same procedure as for 12a. Purification by chromatography on silica gel (1 to 4% methanol in dichloromethane) gave compound 12b (88 mg, 37%). 1H NMR (400 MHz, CDCl3): δ 1.41 (s, 3H, CH3 isop), 1.46 (s, 9H, CH3 Boc), 1.56-1.66 (m, 2H, CH2), 1.64 (s, 3H, CH3 isop), 1.67-1.79 (m, 2H, CH2), 2.05 (s, 3H, COCH3), 2.10 (s, 3H, COCH3), 2.15 (s, 3H, COCH3), 3.15-3.25 (m, 2H, CH2NHBoc), 3.60-3.70 (m, 2H, CH2NH-6), 3.85 (dd, J = 5.2 Hz, J = 10.1 Hz, 1H, H-5’B), 3.92 (dd, J = 4.2 Hz, J = 10.1 Hz, 1H, H-5”B), 4.33 (dd, 1H, J = 6.0 Hz, J = 11.8 Hz, H-5”A), 4.38-4.44 (m, 1H, H-4”A), 4.48 (d, J = 1.0 Hz, 2H, CH2C≡), 4.50-4.55 (m, 2H, H-4’B and H-5”A), 5.10 (dd, J = 3.0 Hz, J = 6.2 Hz, 1H, H-3’B), 5.41 (dd, J = 2.2 Hz, J = 6.2 Hz, 1H, H-2’B), 5.93 (br, 2H, NH2), 5.96 (pt, 1H, J = 6.0 Hz, H-3’A), 6.18 (d, J = 2.2 Hz, 1H, H-1’B), 6.23 (d, 1H, J = 4.2 Hz, H-1’A), 6.30 (dd, J = 4.2 Hz, J = 6.0 Hz, 1H, H-2’A), 7.96 (s, 1H, H-8B), 8.39 (s, 1H, H-2A), 8.43 (s, 1H, H-2B); 13C NMR (100 MHz, CDCl3): δ 20.44 (COCH3), 20.51 (COCH3), 20.65 (COCH3), 25.36 (CH3 isop), 27.04 (CH2), 27.16 (CH3 isop), 27.26 (CH2), 28.44 (3 CH3 Boc), 40.03 (CH2NH-6 and CH2NHBoc), 59.05 (C≡C-CH2), 63.05 (C-5’A), 70.50 (C-3’A), 70.56 (C-5’B), 72.50 (C-2’A), 75.62 (C≡C-CH2), 79.16 (Cq Boc), 79.88 (C-4’A), 81.86 (C-3’B), 84.59 (C-2’B), 85.79 (C-4’B), 87.41 (C-1’A), 91.05 (C-1’B), 91.91 (C≡C-CH2), 114.37 (Cq isop), 118.13 (C-5A); 119.80 (C-5B), 133.42 (C-8A), 138.71 (C-8B), 148.92 (C-4B); 149.40 (C-4A); 153.43 (C-2B); 154.27 (C-2A); 154.96 (C-6B), 155.38 (C-6A), 156.05 (CO Boc), 169.37 (CO), 169.48 (CO), 170.55 (CO); HRMS (ESI-TOF): m/z calcd for [C41H53N11O13+H]+ 908.3803, found 908.4268.
8-[3-(N6-2-Aminoethyl-5’-O-adenosinyl)propargyl]adenosine (2). To a solution of 12a (46 mg, 0.05 mmol) in MeOH (5 mL) was added 28% NH4OH (1 mL). After stirring overnight at room temperature, the reaction mixture was concentrated under reduced pressure, and the crude product (37 mg) was used in the next step without further purification. To the foam (37 mg, 0.05 mmol) was added at 0 °C a 70% aqueous solution of TFA (1 mL). After stirring for 2 h, water was added and the mixture was lyophilized. Purification by reverse phase HPLC (0 to 20% acetonitrile in 10 mM TEAA buffer over 15 min, tR = 13.3 min) afforded compound 2 as acetate salt (13 mg, 42% in two steps). 1H NMR (400 MHz, DMSO-d6): δ 1.88 (s, 1.3H, CH3 Ac), 2.81-2.86 (m, 2H, CH2NH2), 3.54 (d, J = 4.2 Hz, J = 12.2 Hz, 1H, H-5’A), 3.59 (br, 2H, CH2NH-6), 3.68 (d, J = 3.9 Hz, J = 12.2 Hz, 1H, H-5’’A), 3.76 (d, J = 5.5 Hz, J = 10.6 Hz, 1H, H-5’B), 3.85 (dd, J = 3.6 Hz, J = 10.6 Hz, 1H, H-5”B), 3.97-4.02 (m, 1H, H-4’A), 4.08-4.13 (m, 1H, H-4’B), 4.19-4.23 (m, 2H, H-3’A and H-3’B), 4.60 (s, 2H, CH2C≡), 4.63 (t, 1H, J = 5.1 Hz, H-2’B), 5.01 (dd, J = 5.3 Hz, J = 6.6 Hz, 1H, H-2’A), 5.93 (d, 1H, J = 5.1 Hz, H-1’B), 5.95 (d, J = 6.6 Hz, 1H, H-1’A), 7.63 (br s, 2H, NH2), 7.73 (br s, 2H, NH2), 8.17 (s, 1H, H-2B), 8.24 (s, 1H, H-2A), 8.33 (s, 1H, H-8B); 13C NMR (100 MHz, DMSO-d6): δ 21.83 (CH3 Ac), 40.94 (CH2NH2 and CH2NH-6), 58.76 (C≡C-CH2), 62.63 (C-5’A), 70.74 (C-5’B), 71.00 (C-3’B), 71.43 (C-3’A), 72.11 (C-2’A), 73.71 (C-2’B), 75.55 (C≡C-CH2), 83.33 (C-4’B), 87.16 (C-4’A), 87.98 (C-1’B), 89.92 (C-1’A), 92.60 (C≡C-CH2), 119.83 (C-5A and C-5B), 133.45 (C-8A), 139.76 (C-8B), 148.89 (C-4A and C-4B), 153.07 (C-2A), 153.90 (C-2B), 156.63 (C-6A and C-6B), 172.70 (CO Ac); HRMS (ESI-TOF): m/z calcd for [C25H31N11O8+H]+ 614.2435, found 614.2433.
8-[3-(N6-4-Aminobutyl-5’-O-adenosinyl)propargyl]adenosine (3). Compound 3 was obtained from 12b (63 mg, 0.07 mmol) following the same procedure as for 2. Purification by reverse phase HPLC (0 to 30% acetonitrile in 10 mM TEAA buffer over 20 min, tR = 8.7 min) afforded compound 3 as acetate salt (22.6 mg, 50% in two steps). 1H NMR (400 MHz, DMSO-d6): δ 1.48-1.58 (m, 2H, CH2), 1.58-1.69 (m, 2H, CH2), 1.84 (s, 1H, CH3 Ac), 2.73 (t, J = 7.2 Hz, 2H, CH2NH2), 3.50 (br, 2H, CH2NH-6), 3.54 (dd, J = 4.2 Hz, J = 12.1 Hz, 1H, H-5’A), 3.69 (m, J = 3.9 Hz, J = 12.2 Hz, 1H, H-5”A), 3.76 (dd, J = 5.5 Hz, J = 10.5 Hz, 1H, H-5’B), 3.85 (dd, J = 3.8 Hz, J = 10.5 Hz, 1H, H-5”B), 3.98-4.01 (m, 1H, H-4’A), 4.08-4.12 (m, 1H, H-4’B), 4.19-4.23 (m, 2H, H-3’A and H-3’B), 4.60 (s, 2H, CH2C≡), 4.63 (pt, J = 5.3 Hz, 1H, H-2’B), 5.00 (dd, J = 5.3 Hz, J = 6.7 Hz, 1H, H-2’A), 5.93 (d, J = 5.4 Hz, 1H, H-1’B), 5.95 (d, J = 6.7 Hz, 1H, H-1’A), 7.63 (br s, 2H, NH2), 7.79 (br s, 2H, NH2), 8.17 (s, 1H, H-2B), 8.23 (s, 1H, H-2A), 8.32 (s, 1H, H-8B); 13C NMR (100 MHz, DMSO-d6): δ 23.40 (CH3 Ac), 25.81 (CH2), 26.53 (CH2), 39.39 (CH2NH2 and CH2NH-6), 58.73 (C≡C-CH2), 62.49 (C-5’A), 70.66 (C-5’B), 70.83 (C-3’B), 71.29 (C-3’A), 72.12 (C-2’A), 73.56 (C-2’B), 75.51 (C≡C-CH2), 83.33 (C-4’B), 87.07 (C-4’A), 87.82 (C-1’B), 89.88 (C-1’A), 92.72 (C≡C-CH2), 119.71 (C-5A and C-5B), 133.45 (C-8A), 139.76 (C-8B), 148.83 (C-4A and C-4B), 153.18 (C-2A), 153.88 (C-2B), 156.40 (C-6A and C-6B), 173.90 (CO Ac); HRMS (ESI-TOF): m/z calcd for [C27H35N11O8 + H]+ 642.2748, found 642.2748.
5’-[4-(N-tert-Butyloxycarbonyl-amino)butylamido]-5’-deoxy-2’,3’-O-isopropylidene-adenosine (14). To a solution of 5’-amino-5’-deoxy-2’,3’-
O-isopropylidene-adenosine (
13)
[24] (0.31 g, 1.0 mmol) in DMF were added DIEA (0.35 mL, 2.0 mmol), PyBOP (0.52 g, 1.0 mmol) and 4-(
tert-butoxycarbonylamino)-butyric acid (0.20 g, 1.0 mmol). After stirring for 1 h at room temperature, the reaction was diluted with water (40 mL) and extracted with ethyl acetate (2x 80 mL). The combined organic layers were dried over Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography (18 g SiO
2, 0 to 5% methanol in dichloromethane) to yield compound
3 (0.36 g, 74%) as a white foam.
1H NMR (400 MHz, DMSO-
d6):
δ 1.32 (s, 3H, CH
3 isop), 1.36 (s, 9H, CH
3 Boc), 1.54 (s, 3H, CH
3 isop), 1.59 (quint,
J = 7.2 Hz, 2H, CH
2), 2.09 (td,
J = 2.4 Hz,
J = 7.2 Hz, 2H,
CH2CO), 2.89 (br q,
J = 6.6 Hz, 2H,
CH2NHBoc), 3.30-3.35 (m, 2H, H-5’ and H5’’), 4.17 (td,
J = 3.2 Hz,
J = 5.7 Hz, 1H, H-4’), 4.90 (dd,
J = 6.3 Hz,
J = 3.2 Hz, 1H, H-3’), 5.43 (dd,
J = 3.0 Hz,
J = 6.3 Hz, 1H, H-2’), 6.12 (d,
J = 3.0 Hz, 1H, H-1’), 6.75 (br s, 1H, NHBoc), 7.34 (s, 2H, NH
2) 8.06 (br, 1H, NHCO), 8.19 (s, 1H, H-2), 8.32 (s, 1H, H-8);
13C NMR (100 MHz, DMSO-
d6):
δ 25.70 (CH
3 isop), 26.15 (CH
2), 27.50 (CH
3 isop), 28.70 (3 CH
3 Boc), 33.14 (
CH2CO), 39.95 (
CH2NHBoc), 41.16 (C-5’), 77.85 (Cq Boc), 82,17 (C-3’), 83.30 (C-2’), 84.63 (C-4’), 89.55 (C-1’), 113.99 (Cq isop), 119.80 (C-5), 140.53 (C-8), 149.25 (C-4), 153.19 (C-2), 156.02 (CO Boc), 156.67 (C-6), 172.68 (CONH); HRMS (ESI-TOF):
m/z calcd for [C
22H
33N
7O
6+H]
+ 492.2570, found 492.2582.
8-Bromo-5’-(4-(N-tert-butyloxycarbonyl-amino)butylamido)-5’-deoxy-2’,3’-O-isopropylidene-adenosine (7). To a solution of 14 (0.17 g, 0.35 mmol) in 1,4-dioxane (1.4 mL) and sodium acetate buffer (2.1 mL, 0.5 M, pH = 5.3) was added bromine (36 µL, 0.69 mmol) dropwise. After stirring for 3 h at room temperature, the reaction was quenched by adding a saturated aqueous solution of Na2S2O3 (10 mL). After full discoloration, ethyl acetate (80 mL) was added and the organic layer was washed with water (20 mL) and brine (20 mL). The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography (10 g SiO2, 0-4% methanol in dichloromethane) to yield compound 7 (0.135 g, 69%) as a pale yellow foam. 1H NMR (400 MHz, CDCl3): δ 1.38 (s, 3H, CH3 isop), 1.42 (s, 9H, CH3 Boc), 1.65 (s, 3H, CH3 isop), 1.83-1.92 (m, 2H, CH2), 2.32-2.40 (m, 2H, CH2CO), 3.18-3.23 (m, 2H, CH2NHBoc), 3.37 (dt, J = 3.0 Hz, J = 14.3 Hz, 1H, H-5’), 3.99 (ddd, J = 4.0 Hz, J = 8.0 Hz, J = 14.3 Hz, 1H, H-5’’), 4.46 (pq, J = 3.2 Hz, 1H, H-4’), 4.86 (br s, 1H, NHBoc), 4.89 (dd, J = 2.6 Hz, J = 6.2 Hz, 1H, H-3’), 5.45 (dd, J = 4.2 Hz, J = 6.2 Hz, 1H, H-2’), 6.09 (d, J = 4.2 Hz, 1H, H-1’), 6.40 (br s, 2H, NH2), 7.64 (br s, 1H, NHCO), 8.35 (s, 1H, H-2); 13C NMR (100 MHz, CDCl3): δ 25.41 (CH3 isop), 26.04 (CH2), 27.46 (CH3 isop), 28.38 (3 CH3 Boc), 33.49 (CH2CO), 39.94 (CH2NHBoc), 40.93 (C-5’), 79.22 (Cq Boc), 81.49 (C-3’), 82.18 (C-2’), 83.95 (C-4’), 91.98 (C-1’), 114.83 (Cq isop), 120.51 (C-5), 127.79 (C-8), 150.17 (C-4), 151.59 (C-2), 153.96 (C-6), 156.23 (CO Boc), 173.01 (CONH); HRMS (ESI-TOF): m/z calcd for [C22H32BrN7O6+H]+ 570.1675, found 570.1683.
5’-O-[3-(5’-(4-(N-tert-Butyloxycarbonyl-amino)butylamido)-5’-deoxy-2’,3’-O-isopropylideneadenosine-8-yl)prop-2-yn-1-yl]-2’,3’-O-isopropylideneadenosine (15). To a solution of bromide 7 (0.19 g, 0.34 mmol) and alkyne 8c [
18] (0.18 g, 0.51 mmol) in THF (4.0 mL) was added NEt
3 (0.14 mL, 1.02 mmol). The stirred mixture was degassed by vacuum/argon purging (3 times) before adding sequentially CuI (7 mg, 0.036 mmol) and Pd(PPh
3)
4 (20 mg, 0.017 mmol), followed by another 15 min argon degassing. After stirring for 3 h at 60 °C, reaction was incomplete (TLC monitoring), Pd(PPh
3)
4 (40 mg, 0.034 mmol) was added. After 3 h at 60 °C, volatiles were removed and the residue was purified by flash column chromatography (18 g SiO
2, 0 to 7% methanol in dichloromethane) to yield 15 (74 mg, 25 %) as a pale orange foam.
1H NMR (400 MHz, DMSO-d
6): δ 1.28 (s, 3H, CH
3 isop), 1.34 (s, 3H, CH
3 isop), 1.35 (s, 9H, CH
3 Boc), 1.48 (s, 3H, CH
3 isop), 1.55 (s, 3H, CH
3 isop), 1.58 (quint, J = 7.2 Hz, 2H, CH
2), 2.04-2.08 (m, 2H, CH
2CO), 2.89 (q, J = 6.6 Hz, 2H, CH
2CO), 3.30-3.40 (m, 2H, H-5’A and H-5”A), 3.72 (dd, J = 5.0 Hz, J = 10.3 Hz, 1H, H-5’B), 3.80 (dd, J = 5.7 Hz, J = 10.3 Hz, 1H, H-5’’B), 4.14-4.18 (m, 1H, H-4’A), 4.36-4.40 (m, 1H, H-4’B), 4.58 (s, 2H, CH
2C≡), 4.95 (dd, J = 3.4 Hz, J = 6.3 Hz, 1H, H-3’A), 5.02 (dd, J = 3.0 Hz, J = 6.2 Hz, 1H, H-3’B), 5.43 (dd, J = 2.6 Hz, J = 6.2 Hz, 1H, H-2’B), 5.51 (dd, J = 2.7 Hz, J = 6.3 Hz, 1H, H-2’A), 6.11 (d, J = 2.7 Hz, 1H, H-1’A), 6.19 (d, J = 2.6 Hz, 1H, H-2’B), 6.73 (br, 1H, NHBoc), 7.54 (br, 2H, NH
2), 7.65 (br, 2H, NH
2), 8.03 (t, J = 5.7 Hz, 1H, NHCO), 8.22 (s, 1H, H-2B, 8.24 (s, 1H, H-2A), 8.36 (s, 1H, H-8B);
13C NMR (100 MHz, DMSO-d
6): δ 25.66 (2 CH
3 isop), 26.12 (CH
2), 27.50 (2 CH
3 isop), 28.69 (3 CH
3 Boc), 33.12 (CH
2CO), 39.82 (CH
2NHBoc), 41.06 (C-5’A), 58.67 (C≡C-CH
2), 70.33 (C-5’B), 75.50 (C≡C-CH
2), 77.85 (Cq Boc), 81.91 (C-3’B), 82.37 (C-3’A), 82.96 (C-2’A), 83.80 (C-2’B), 84.81 (C-4’B), 85.25 (C-4’A), 89.76 (C-1’A and C-1’B), 92.75 (C≡C-CH
2), 113.90 (Cq isop), 114.01 (Cq isop), 119.44 (C-5A and C-5B), 132.57 (C-8A), 140.45 (C-8B), 148.78 (C-4A), 149.24 (C-4B), 152.24 (C-2A), 154.24 (C-2B), 155.76 (C-6B), 156.03 (C-6A), 156.38 (CO Boc), 172.60 (CONH); HRMS (ESI-TOF):
m/z calcd for [C
38H
50N
12O
10+H]
+ 835.3851, found 835.3840.
5’-O-[3-(5’-(4-Aminobutylamido)-5’-deoxy-adenosin-8-yl)prop-2-yn-1-yl]-adenosine (4). Compound 15 (64 mg, 0.08 mmol) was dissolved in an ice-cold solution of 50% aqueous TFA (5.0 mL) and the reaction mixture was allowed to warm to room temperature. After stirring for 3 h at room temperature, water was added and the reaction mixture was lyophilized. Purification of the residue by reverse phase HPLC (5 to 25% acetonitrile in 10 mM TEAA buffer over 15 min) yielded 4 (22.6 mg, 45%) as acetate salt. tR = 10.4 min; 1H NMR (400 MHz, DMSO-d6): δ 1.68 (quint, J = 7.2 Hz, 2H, CH2), 1.79 (s, 1.88 H, CH3 Ac), 2.20 (t, J = 6.2 Hz, 2H, CH2CO), 2.65 (t, J = 6.2 Hz, 2H, CH2NH2), 3.38 (dt, J = 4.8 Hz, J = 14.0 Hz, 1H, H-5’A), 3.47 (dt, J = 5.8 Hz, J = 14.0 Hz, 1H, H-5”A), 3.77 (dd, J = 5.6 Hz, J = 10.6 Hz, 1H, H-5’B), 3.85 (dd, J = 3.7 Hz, J = 10.6 Hz, 1H, H-5”B), 3.98-4.01 (m, 1H, H-4’A), 4.08-4.13 (m, 2H, H-3’A and H-4’B), 4.20 (t, J = 4.7 Hz, 1H, H-3’B), 4.61-4.63 (m, 3H, H-2’B and CH2C≡), 5.01-5.04 (m, 1H, H-2’A), 5.92 (d, J = 5.3 Hz, 1H, H-1’B), 5.94 (d, J = 6.4 Hz, 1H, H-1’A), 7.25 (s, 2H, NH2), 7.62 (s, 2H, NH2), 8.17 (s, 1H, H-2B), 8.24 (s, 1H, H-2A), 8.27 (m, 1H, NHCO), 8.32 (s, 1H, H-8B); 13C NMR (100 MHz, DMSO-d6): δ 23.08 (CH3 Ac), 26.58 (CH2), 33.02 (CH2CO), 40.02 (CH2NH2), 41.27 (C-5’A), 58.74 (C≡C-CH2), 70.77 (C-5’B), 71.02 (C-3’B), 71.71 (C-3’A and C-2’A), 73.69 (C-2’B), 75.70 (C≡C-CH2), 83.28 (C-4’B), 84.55 (C-4’A), 87.96 (C-1’B), 89.67 (C-1’A), 92.56 (C≡C-CH2), 119.50 (C-5B), 119.81 (C-5A), 133.41 (C-8A), 139.84 (C-8B), 149.16 (C-4A), 149.95 (C-4B), 153.18 (C-2B), 154.21 (C-2A), 156.50 (C-6B), 156.59 (C-6A), 172.41 (CONH), 173.60 (CO Ac); HRMS (ESI-TOF): m/z calcd for [C27H34N12O8+H]+ 655.2701, found 655.2708.
5’-Deoxy-5’-(2-nitrobenzenesulfonamido)-2’,3’-O-isopropylidene-adenosine (16). To a solution of 13 (2.30 g, 7.5 mmol) in pyridine (75 mL) was added 2-nitro-benzenesulfonyl chloride (3.82 g, 17.25 mmol). After stirring for 2 h at room temperature, volatiles were removed and the residue was purified by flash column chromatography (240 g SiO2, 3% isocratic methanol in dichloromethane) to yield compound 16 (3.25 g, 88%) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 1.29 (s, 3H, CH3 isop), 1.52 (s, 3H, CH3 isop), 3.19-3.33 (m, 2H, H-5’ and H-5’’), 4.24 (td, J = 2.9 Hz, J = 5.7 Hz, 1H, H-4’), 4.95 (dd, J = 2.9 Hz, J = 6.2 Hz, 1H, H-3’), 5.37 (dd, J = 2.9 Hz, J = 6.2 Hz, H-2’), 6.12 (d, J = 2.9 Hz, H-1’), 7.38 (s, 2H, NH2), 7.70-7.74 (m, 1H, H-4 Ns), 7.78-7.82 (m, 1H, H-3 Ns), 7.85-7.87 (m, 1H, H-5 Ns), 7.91-7-93 (m, 1H, H-2 Ns), 8.13 (s, 1H, H-8), 8.29 (s, 1H, H-2), 8.60 (br s, 1H, NH-5’); 13C NMR (100 MHz, DMSO-d6): δ 25.60 (CH3 isop), 27.42 (CH3 isop), 45.12 (C-5’), 82.01 (C-3’), 83.33 (C-2’), 84.68 (C-4’), 90.00 (C-1’), 133.94 (Cq isop), 299.82 (C-5), 124.83 (C-3 Ns), 129.90 (C-6 Ns), 132.93 (C-5 Ns), 133.01 (C-1 Ns), 134.50 (C-4 Ns), 140.63 (C-8), 148.00 (C-2 Ns), 148.90 (C-4), 153.05 (C-2), 156.71 (C-6); HRMS (ESI-TOF): m/z calcd for [C19H21N7O7S+H]+ 492.1302 found, 492.1302.
5’-N-(N-tert-Butyloxycarbonyl-aminopropyl)amino-5’-deoxy-2’,3’-O-isopropylidene-adenosine (17). To a solution of
17 (0.80 g, 1.63 mmol) in DMF was added K
2CO
3 (0.68 g, 4.88 mmol) followed by
tert-butyl-3-bromopropylcarbamate (prepared according reported procedures) [
25] (0.47 g, 1.95 mmol). After stirring overnight at 50 °C, alkylation was incomplete and
tert-butyl-3-bromopropylcarbamate (0.23 g, 0.98 mmol) was further added. After heating at 50 °C for 24 h, thiophenol (0.36 mL, 3.26 mmol) was added and stirring was maintained for 18 h at room temperature. Volatiles were then removed under reduced pressure and the residue purified by flash column chromatography (85 g SiO
2 70–230 mesh, 5 to 11% methanol in dichloromethane to yield compound
17 (0.61 g, 79%) as a white foam.
1H NMR (400 MHz, DMSO-
d6):
δ 1.33 (s, 3H, CH
3 isop), 1.36 (s, 9H, CH
3 Boc), 1.48 (quint,
J = 6.8 Hz, 2H, CH
2), 1.54 (s, 3H, CH
3 isop), 2.46-2.51 (m, 2H,
CH2NH), 2.66 (dd,
J = 6.1 Hz,
J = 12.2 Hz, 2H, H-5’), 2.73 (dd,
J = 5.9 Hz,
J = 12.2 Hz, 2H, H-5”), 2.93 (br q,
J = 6.8 Hz, 2H,
CH2NHBoc), 4.21 (td,
J = 2.8 Hz,
J = 5.9 Hz, 1H, H-4’), 4.97 (dd,
J = 6.3 Hz,
J = 2.8 Hz, 1H, H-3’), 5.45 (dd,
J = 3.0 Hz,
J = 6.3 Hz, 1H, H-2’), 6.09 (d,
J = 3.0 Hz, 1H, H-1’), 6.75 (br, 1H, NHBoc), 7.30 (s, 2H, NH
2), 8.16 (s, 1H, H-8), 8.35 (s, 1H, H-2);
13C NMR (100 MHz, DMSO-
d6):
δ 25.72 (CH
3 isop), 27.53 (CH
3 isop), 28.72 (3 CH
3 Boc), 30.08 (CH
2), 38.51 (
CH2NHBoc), 47.26 (
CH2NH), 51.47 (C-5’), 77.81 (Cq Boc), 82.65 (C-3’), 83.20 (C-2’), 85.33 (C-4’), 89.75 (C-1’), 113.68 (Cq isop), 119.71 (C-5), 140.42 (C-8), 149.41 (C-4), 153.15 (C-2), 156.07 (CO Boc), 156.62 (C-6); HRMS (ESI-TOF):
m/z calcd for [C
21H
33N
7O
5+H]
+ 464.2621, found 464.2628.
5’-N-(N-tert-Butyloxycarbonyl-aminopropyl)propargylamino-5’-deoxy-2’,3’-O-isopropylidene-adenosine (18). To a solution of 17 (0.60 g, 1.3 mmol) in DMF (13 mL), were added DIEA (1.35 mL, 7.7 mmol) and propargyl bromide (80% in toluene, 0.79 mL, 7.10 mmol). After stirring for 2 h at room temperature, the reaction was diluted with ethyl acetate (100 mL) and washed with water (2 x 50 mL). Organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography (20 g SiO2, 3 to 4% methanol in dichloromethane) to yield compound 18 (0.53 g, 82%) as a white foam. 1H NMR (400 MHz, DMSO-d6): δ 1.33 (s, 3H, CH3 isop), 1.37 (s, 9H, CH3 Boc), 1.46 (quint, J = 6.8 Hz, 2H, CH2), 1.54 (s, 3H, CH3 isop), 2.40 (t, J = 6.8 Hz, 2H, CH2N), 2.51-2.55 (m, 1H, H-5’), 2.67 (dd, J = 7.6 Hz, J = 13.1 Hz, 1H, H-5”), 2.84-2.99 (m, 2H, CH2NHBoc), 3.01 (t, J = 2.2 Hz, 1H, HC≡C), 3.29-3.41 (m, 2H, CH2C≡), 4.22 (td, J = 2.8 Hz, J = 6.9 Hz, 1H, H-4’), 4.99 (dd, J = 2.8 Hz, J = 6.3 Hz, 1H, H-3’), 5.49 (dd, J = 2.4 Hz, J = 6.3 Hz, 1H, H-2’), 6.14 (dd, J = 2.4 Hz, 1H, H-1’), 6.72 (br, 1H, NHBoc), 7.30 (s, 2H, NH2), 8.18 (s, 1H, H-8), 8.31 (s, 1H, H-2); 13C NMR (100 MHz, DMSO-d6): δ 25.67 (CH3 isop), 27.43 (CH3 isop), 27.79 (CH2), 28.73 (3 CH3 Boc), 38.39 (CH2NHBoc), 42.40 (C≡C-CH2), 51.53 (CH2N), 55.32 (C-5’), 76.01 (HC≡C-CH2), 77.84 (Cq Boc), 79.21 (C≡C-CH2), 83.34 (C-3’), 83.43 (C-2’), 84.75 (C-4’), 89.67 (C-1’), 113.64 (Cq isop), 119.69 (C-5), 140.47 (C-8), 149.28 (C-4), 153.18 (C-2), 156.04 (CO Boc), 156.60 (C-6); HRMS (ESI-TOF): m/z calcd for [C24H35N7O5+H]+ 502.2778, found 502.2770.
5’-Deoxy-5’-N-(N-tert-Butyloxycarbonyl-aminopropyl)-3-(2’,3’,5’-tri-O-acetyl-adenosine-8-yl)propargyl amino-2’,3’-O-isopropylidene-adenosine (19). To a solution of bromide 6 (0.26 g, 0.55 mmol) and alkyne 18 (0.41 g, 0.82 mmol) in THF (16.5 mL) was added NEt3 (0.23 mL, 1.65 mmol). The stirred mixture was degassed with argon (3 times) before adding sequentially CuI (10 mg, 0.05 mmol) and Pd(PPh3)4 (32 mg, 0.028 mmol), followed by argon degassing. After stirring for 4 h at 60 °C, a second addition of Pd(PPh3)4 (32 mg, 0.028 mmol) was made. After 18 h at 60 °C, crude was filtered over Celite and volatiles were removed under reduced pressure. The residue was purified by flash column chromatography (31 g SiO2, 0 to 7% methanol in dichloromethane) to yield 7 (0.31 g, 63%) as a white foam. 1H NMR (400 MHz, DMSO-d6): δ 1.34 (s, 3H, CH3 isop), 1.37 (s, 9H, CH3 Boc), 1.55 (s, 3H, CH3 isop), 1.51-1.60 (m, 2H, CH2), 1.95 (s, 3H, COCH3), 1.96 (s, 3H, COCH3), 2.06 (s, 3H, COCH3), 2.51-2.56 (m, 2H, CH2N), 2.66 (dd, J = 6.0 Hz, J = 13.2 Hz, 1H, H-5’B), 2.82 (dd, J = 8.0 Hz, J = 13.2 Hz, 1H, H-5”B), 2.89-3.05 (m, 2H, CH2NHBoc), 3.77-3.88 (m, 2H, CH2C≡), 4.16 (dd, J = 5.6 Hz, J = 12.0 Hz, 1H, H-5’A), 4.28-4.35 (m, 2H, H-4’A and H-4’B), 4.41 (dd, J = 3.7 Hz, J = 12.0 Hz, 1H, H-5”A), 5.04 (dd, J = 2.8 Hz, J = 6.3 Hz, 1H, H-3’B), 5.51 (dd, J = 2.5 Hz, J = 6.4 Hz, 1H, H-2’B), 5.72 (t, J = 6.0 Hz, 1H, H-3’A), 6.11 (d, J = 4.6 Hz, 1H, H-1’A), 6.17 (d, J = 2.5 Hz, 1H, H-1’B), 6.19 (dd, J = 4.6 Hz, J = 6.0 Hz, 1H, H-2’A), 6.77 (br t, J = 5.4 Hz, 1H, NHBoc), 7.31 (br, 1H, NH2), 7.57 (s, 2H, NH2), 8.18 (s, 1H, H-2A), 8.19 (s, 1H, H-2B), 8.33 (s, 1H, H-8B); 13C NMR (100 MHz, DMSO-d6): δ 20.48 (CH3), 20.69 (CH3), 20.84 (CH3), 25.66 (CH3 isop), 27.43 (CH3 isop), 27.90 (CH2), 28.70 (3 CH3 Boc), 38.31 (CH2NHBoc), 43.16 (C≡C-CH2), 51.78 (CH2N), 55.35 (C-5’B), 62.86 (C-5’A), 70.24 (C-3’A), 71.71 (C-2’A), 74.34 (C≡C-CH2), 77.90 (Cq Boc), 79.69 (C-4’A), 83.26 (C-3’B), 83.43 (C-2’B), 84.33 (C-4’B), 87.30 (C-1′A), 89.71 (C-1′B), 93.23 (C≡C-CH2), 113.77 (Cq isop), 119.26 (C-5A), 119.71 (C-5B), 133.28 (C-8A), 140.54 (C-8B), 149.04 (C-4B), 149.24 (C-4A), 153.19 (C-2A), 154.32 (C-2B), 156.09 (CO Boc), 156.43 (C-5B), 156.62 (C-6A), 169.73 (CO), 169.81 (CO), 170.41 (CO); HRMS (ESI-TOF): m/z calcd for [C40H52N12O12+H]+ 893.3906, found 893.3939.
5’-[N-3-Aminopropyl-N-(3-(adenosin-8-yl)propargyl)amino]-5’-deoxyadenosine (5). To a solution of 7 (0.29 g, 0.33 mmol) in methanol (3.3 mL) was added 28% NH4OH (0.80 mL, 13.3 mmol). After stirring for 18 h at room temperature, volatiles were removed under reduced pressure and the crude material was brought to 0 °C before adding an ice-cold solution of TFA (50% in water, 10 mL). After 4 h at room temperature, water was added and the reaction mixture was lyophilized. Purification by reverse phase HPLC (0 to 30% acetonitrile in 10 mM TEAA buffer over 15 min) afforded the fully deprotected compound 5 as acetate salt (0.12 g, 61%) as white powder. tR = 10.8 min; 1H NMR (400 MHz, DMSO-d6): δ 1.65 (quint, J = 6.8 Hz, 2H, CH2), 1.78 (s, 3H, CH3 Ac), 2.65 (t, J = 6.8 Hz, 2H, CH2N), 2.70 (t, J = 6.8 Hz, 2H, CH2NH2), 2.80 (dd, J = 4.8 Hz, J = 13.7 Hz, 1H, H-5’B), 2.90 (dd, J = 6.8 Hz, J = 13.7 Hz, 1H, H-5”B), 3.53 (dd, J = 4.0 Hz, J = 12.2 Hz, 1H, H-5’A), 3.69 (dd, J = 3.6 Hz, J = 12.2 Hz, 1H, H-5”A), 3.80 (s, 2H, CH2C≡), 3.96-4.01 (m, 1H, H-4’A), 4.03-4.08 (m, 1H, H-4’B), 4.14 (pt, J = 5.0 Hz, 1H, H-3’B), 4.20 (dd, J = 2.3 Hz, J = 5.1 Hz, 1H, H-3’A), 4.67 (pt, J = 5.1 Hz, 1H, H-2’B), 4.98 (dd, J = 5.3 Hz, J= 6.0 Hz, 1H, H-2’A), 5.89 (d, J = 5.4 Hz, 1H, H-1’B), 6.00 (dd, J = 6.8 Hz, H-1’A), 7.25 (br, 1H, NH2 B), 7.61 (s, 2H, NH2 A), 8.15 (s, 1H, H-2A), 8.17 (s, 1H, H-2B), 8.36 (s, 1H, H-8B); 13C NMR (100 MHz, DMSO-d6): δ 23.23 (CH3 Ac), 28.50 (CH2N), 38.89 (CH2NH2), 44.80 (C≡C-CH2), 51.78 (CH2N), 56.23 (C-5’B), 62.69 (C-5’A), 71.49 (C-3’A), 72.21 (C-3’B and C-2’A), 73.14 (C-2’B), 74.81 (C≡C-CH2), 83.03 (C-4’B), 87.11 (C-4’A), 88.10 (C-1′B), 90.01 (C-1′A), 92.96 (C≡C-CH2), 119.64 and 119.67 (C-5A and C-5B), 133.94 (C-8A), 140.24 (C-8B), 148.84 (C-4A), 149.90 (C-4B), 153.14 (C-2B), 153.67 (C-2A), 156.52 (C-6B), 156.54 (C-6A), 173.90 (CO Ac); HRMS (ESI-TOF): m/z calcd for [C26H34N12O7+H]+ 627.2751 found, 627.2731.


 ,
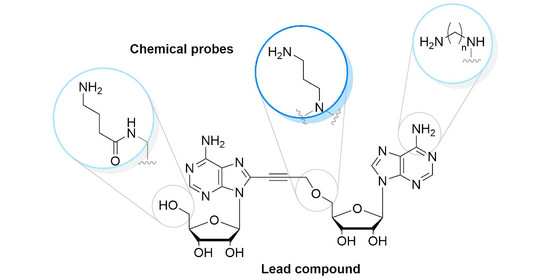
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







