
Monitoring the Diversity and Metabolic Shift of Gut Microbes during Green Tea Feeding in an In Vitro Human Colonic Model

_Zhu.png)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Human Colon Microbiota
2.3. In Vitro Human Colonic Model (HCM)
2.4. Green Tea Extraction
2.5. 16 S rRNA Gene Library Preparation and Analysis
2.6. Metabolites Extraction
2.7. Polyphenol Extraction
2.8. Targeted HPLC-MS/MS Metabolic Profiling
2.9. Targeted HPLC-MS/MS Phenolic Compounds Profiling
2.10. Statistical and Bioinformatics Analysis
3. Results and Discussion
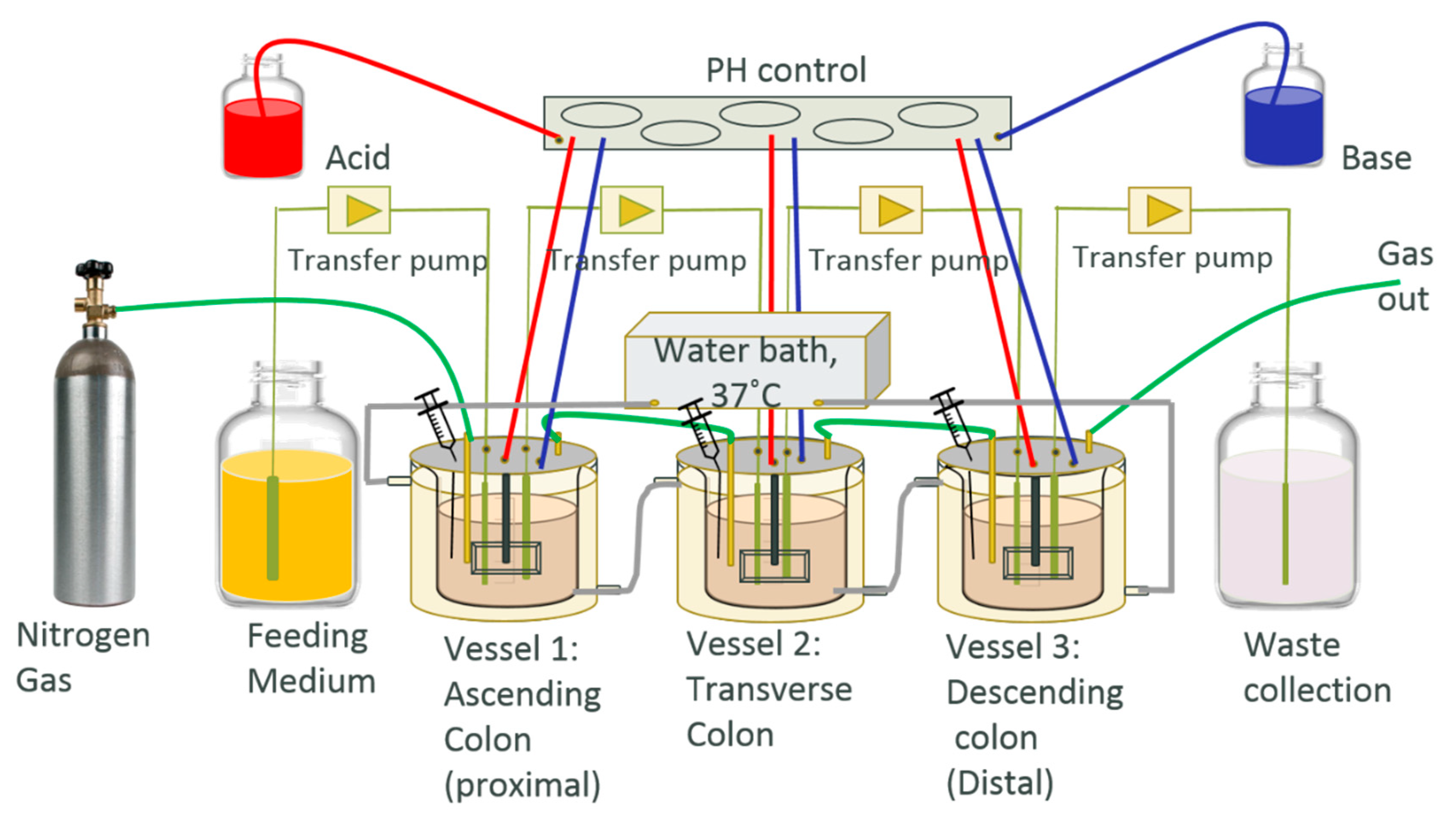
3.1. Design of Human Colonic Model (HCM) System
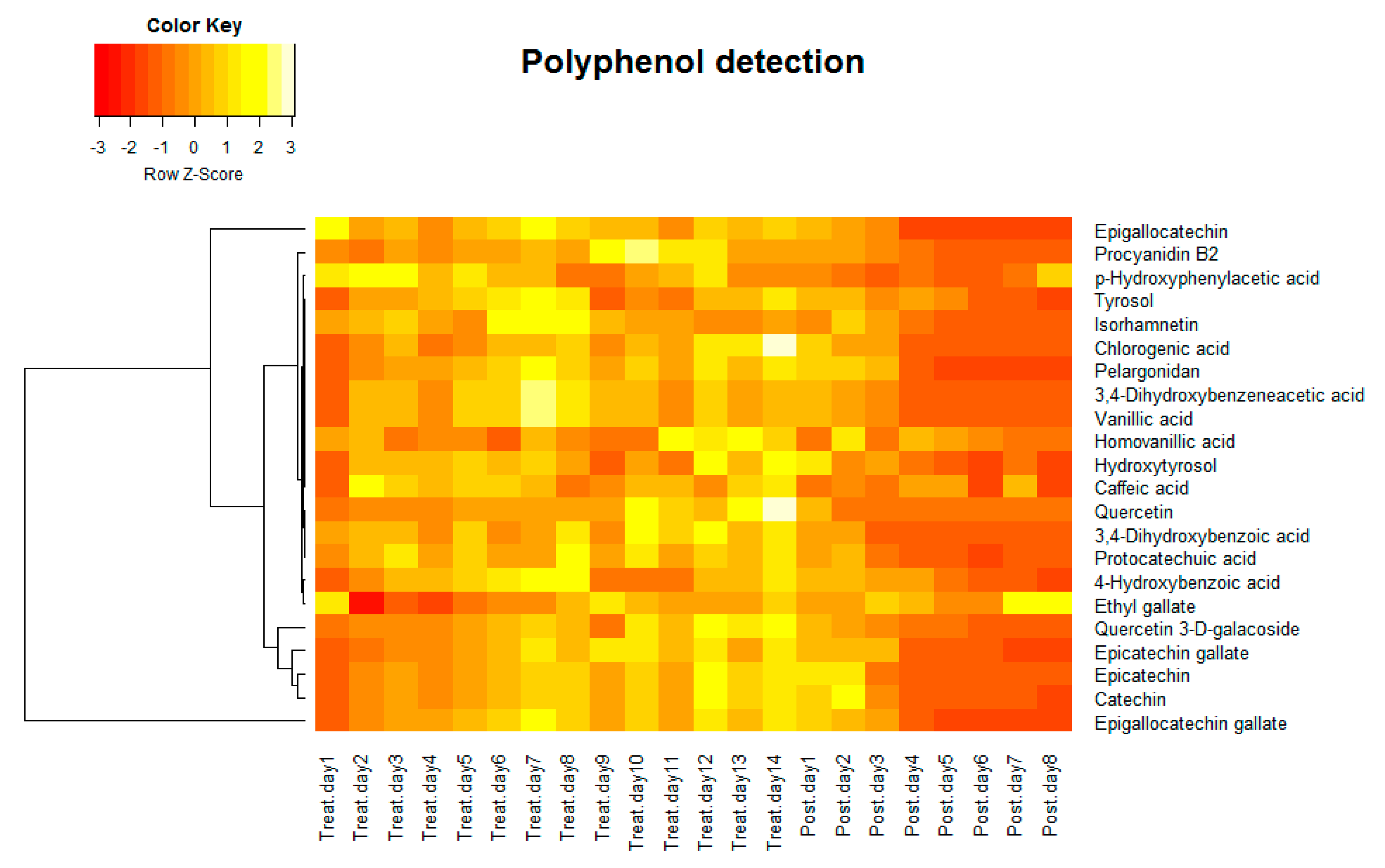
3.2. Tracking the Phenolic Compounds Contents in Gut Microbiota
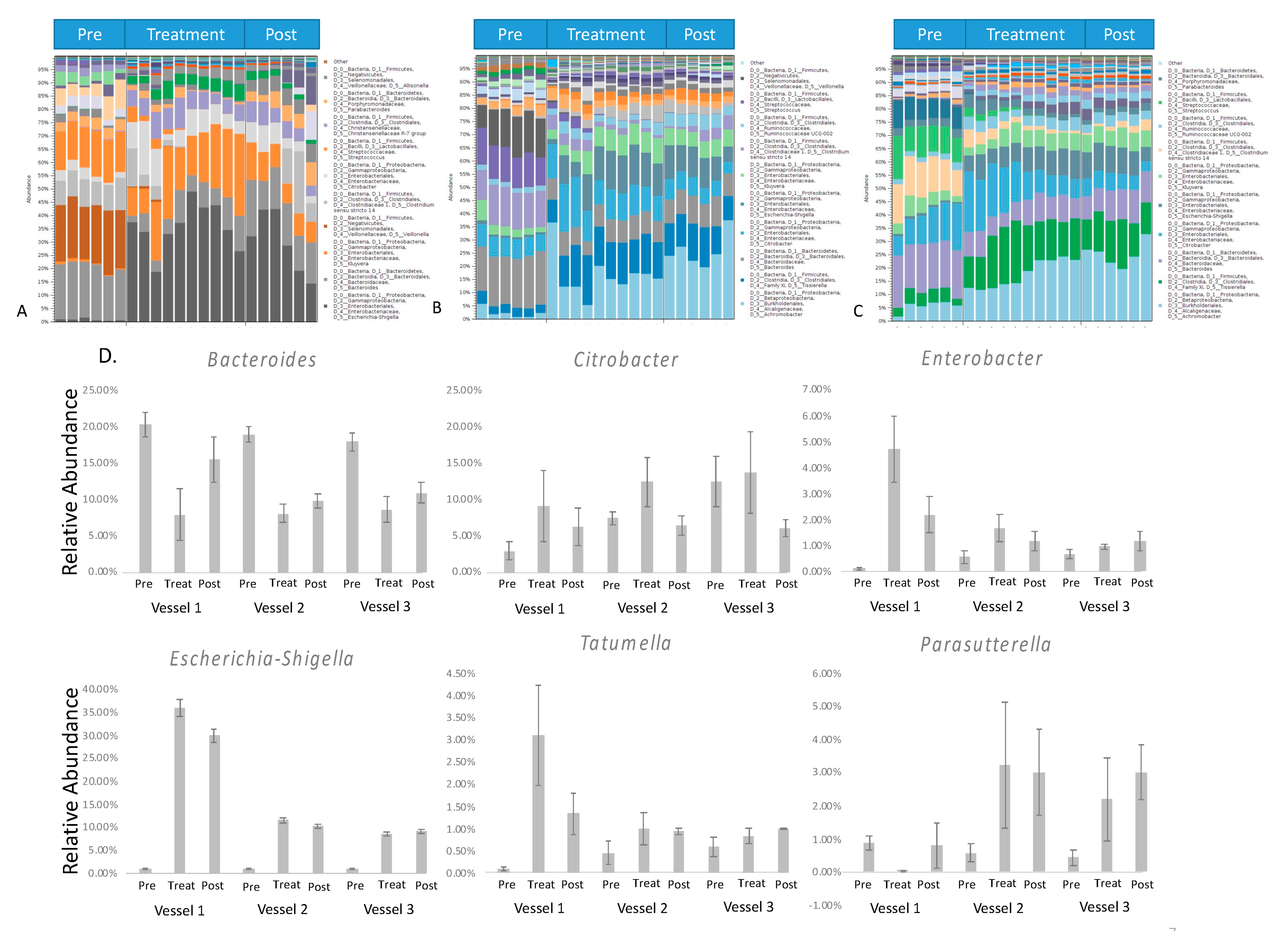
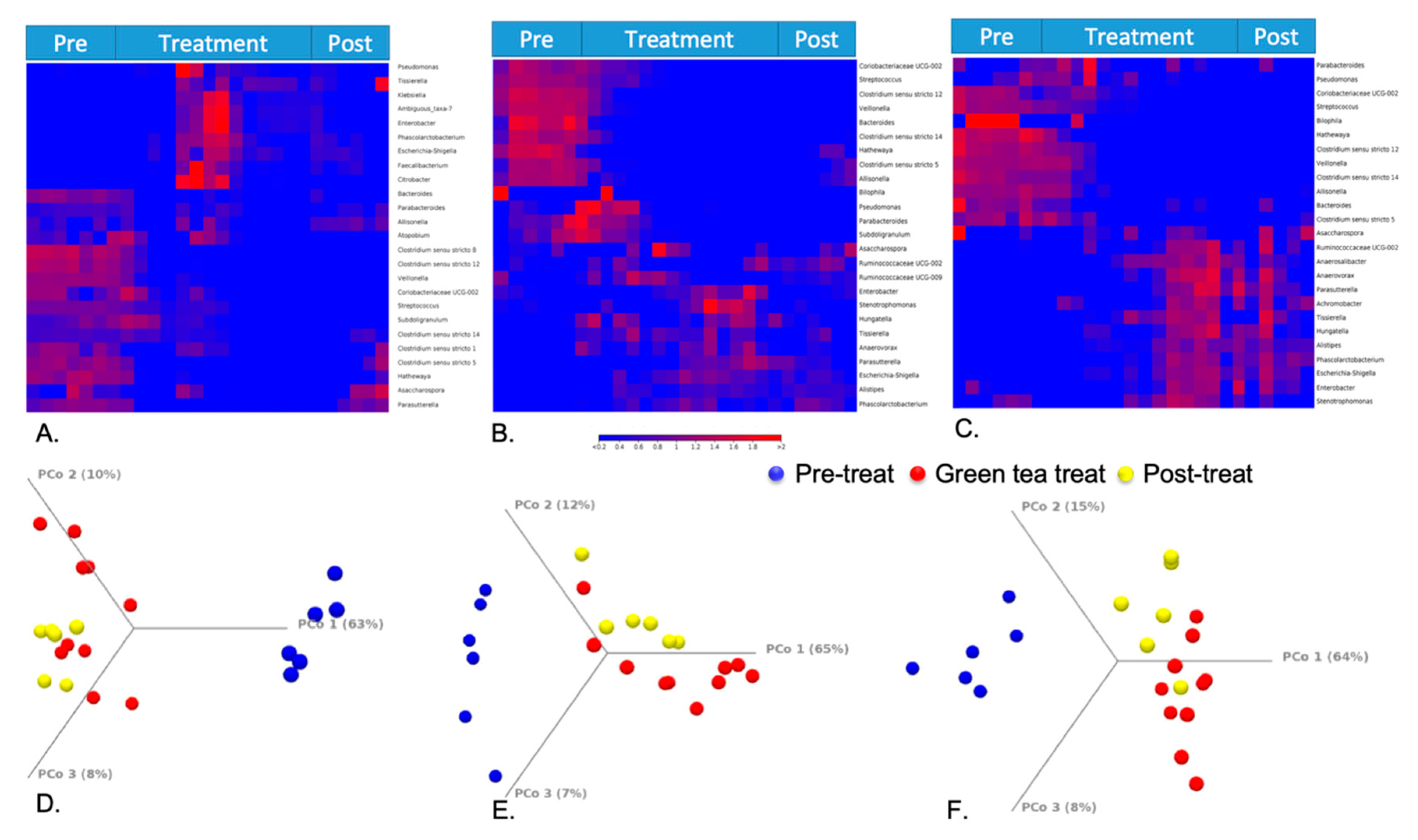
3.3. Microbial Composition Changes in Response to Green Tea Polyphenols
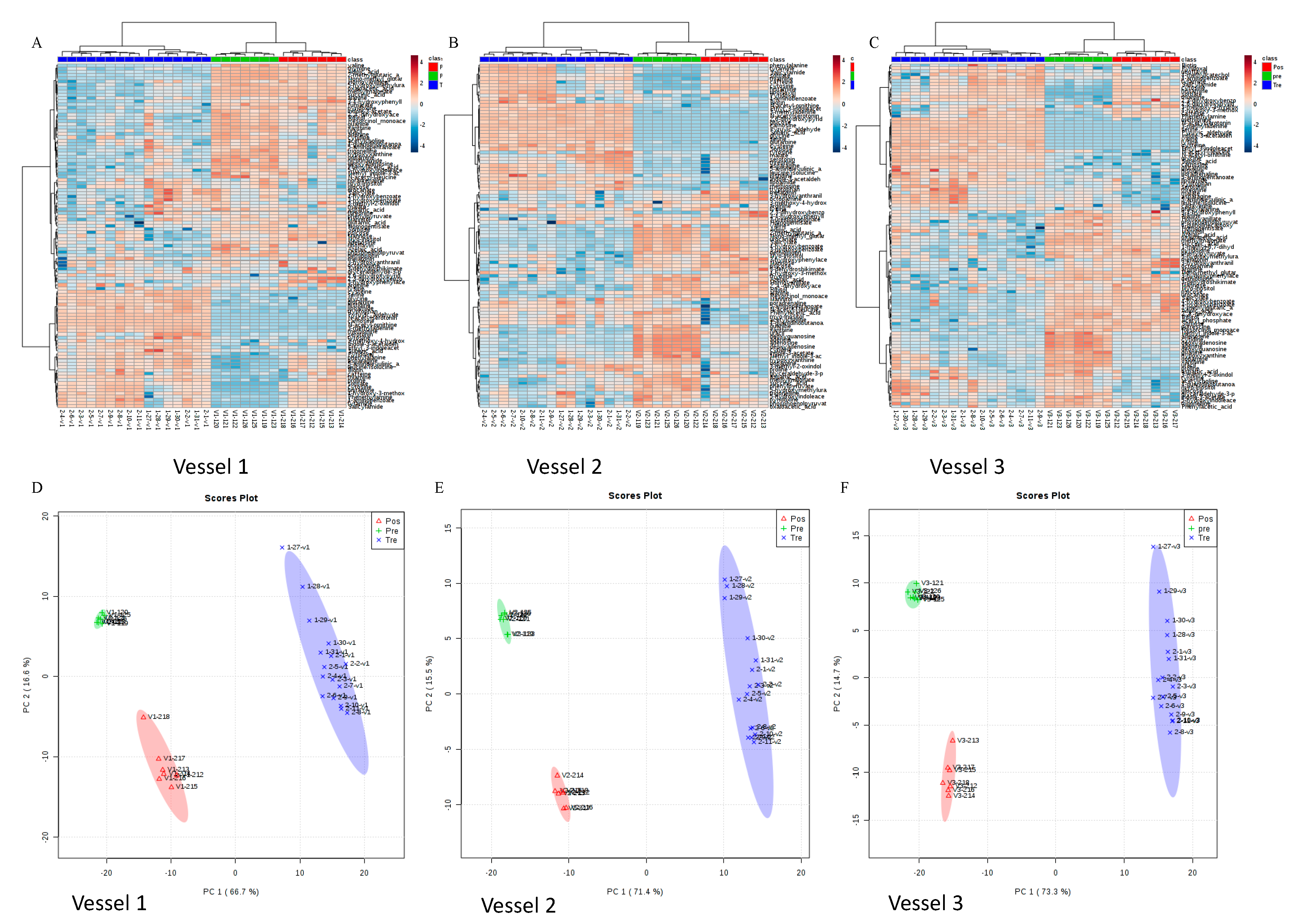
3.4. Green Tea Polyphenol-Induced Gut Microbial Metabolic Changes and Their Association with Microbial Composition Changes
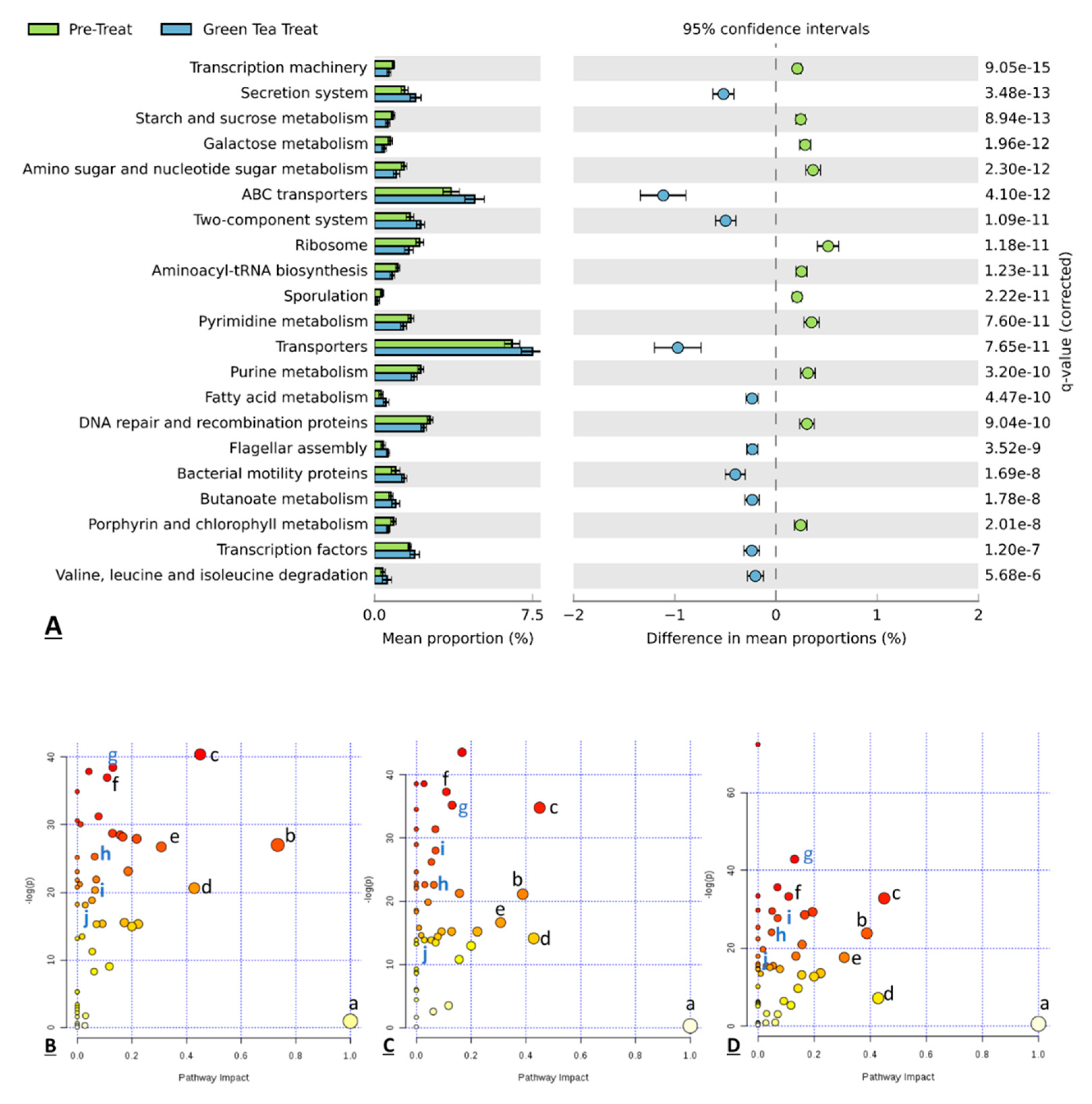
3.5. Functional Prediction of the Gut Microbiota
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, B.; Yao, M.; Lv, L.; Ling, Z.; Li, L. The human microbiota in health and disease. Engineering 2017, 3, 71–82. [Google Scholar] [CrossRef]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70, S38–S44. [Google Scholar] [CrossRef] [Green Version]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, K.B.; Leone, V.A.; Chang, E.B. Microbial metabolites in health and disease: Navigating the unknown in search of function. J. Biol. Chem. 2017, 292, 8553–8559. [Google Scholar] [CrossRef] [Green Version]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [Green Version]
- Gibson, G.; Rastall, R.; Fuller, R. The health benefits of probiotics and prebiotics. Gut FloraNutr. Immun. Health 2003, 52–76. [Google Scholar]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, ra14–ra16. [Google Scholar] [CrossRef] [Green Version]
- Esposito, E.; Iacono, A.; Bianco, G.; Autore, G.; Cuzzocrea, S.; Vajro, P.; Canani, R.B.; Calignano, A.; Raso, G.M.; Meli, R. Probiotics reduce the inflammatory response induced by a high-fat diet in the liver of young rats. J. Nutr. 2009, 139, 905–911. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Hua, J.; Li, Z. Probiotics improve high fat diet-induced hepatic steatosis and insulin resistance by increasing hepatic NKT cells. J. Hepatol. 2008, 49, 821–830. [Google Scholar] [CrossRef] [Green Version]
- Axling, U.; Olsson, C.; Xu, J.; Fernandez, C.; Larsson, S.; Ström, K.; Ahrné, S.; Holm, C.; Molin, G.; Berger, K. Green tea powder and Lactobacillus plantarum affect gut microbiota, lipid metabolism and inflammation in high-fat fed C57BL/6J mice. Nutr. Metab. 2012, 9, 105. [Google Scholar] [CrossRef] [Green Version]
- Gradišar, H.; Pristovšek, P.; Plaper, A.; Jerala, R. Green Tea Catechins Inhibit Bacterial DNA Gyrase by Interaction with Its ATP Binding Site. J. Med. Chem. 2007, 50, 264–271. [Google Scholar] [CrossRef]
- Mujtaba, T.; Dou, Q.P. Black Tea Polyphenols Inhibit Tumor Proteasome Activity. Vivo (AthensGreece) 2012, 26, 197–202. [Google Scholar]
- Ruxton, C.H.S. The health effects of black tea and flavonoids. Nutr. Food Sci. 2009, 39, 283–294. [Google Scholar] [CrossRef]
- Lambert, J.D.; Elias, R.J. The antioxidant and pro-oxidant activities of green tea polyphenols: A role in cancer prevention. Arch. Biochem. Biophys. 2010, 501, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Senanayake, S.P.J.N. Green tea extract: Chemistry, antioxidant properties and food applications-A review. J. Funct. Foods 2013, 5, 1529–1541. [Google Scholar] [CrossRef]
- Yang, F.J.; de Villers, W.J.S.; McClain, C.J.; Varilek, G.W. Green tea polyphenols block endotoxin-induced tumor necrosis factor-production and lethality in a murine model. J. Nutr. 1998, 128, 2334–2340. [Google Scholar] [CrossRef]
- Shivashankara, K.; Acharya, S. Bioavailability of dietary polyphenols and the cardiovascular diseases. Open Nutraceuticals J. 2010, 3, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Aziz, N.; Farag, S.; Mousa, L.; Abo-Zaid, M. Comparative antibacterial and antifungal effects of some phenolic compounds. Microbios 1998, 93, 43–54. [Google Scholar]
- Lee, J.-H.; Shim, J.S.; Lee, J.S.; Kim, J.K.; Yang, I.S.; Chung, M.-S.; Kim, K.H. Inhibition of pathogenic bacterial adhesion by acidic polysaccharide from green tea (Camellia sinensis). J. Agric. Food Chem. 2006, 54, 8717–8723. [Google Scholar] [CrossRef]
- Ankolekar, C.; Johnson, D.; Pinto, M.d.S.; Johnson, K.; Labbe, R.; Shetty, K. Inhibitory potential of tea polyphenolics and influence of extraction time against Helicobacter pylori and lack of inhibition of beneficial lactic acid bacteria. J. Med. Food 2011, 14, 1321–1329. [Google Scholar] [CrossRef]
- Ahn, Y.; Sakanaka, S.; Kim, M.; Kawamura, T.; Fujisawa, T.; Mitsuoka, T. Effect of green tea extract on growth of intestinal bacteria. Microb. Ecol. Health Dis. 1990, 3, 335–338. [Google Scholar]
- McGhie, T.K.; Walton, M.C. The bioavailability and absorption of anthocyanins: Towards a better understanding. Mol. Nutr. Food Res. 2007, 51, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.P.E. Metabolism of Tea Flavonoids in the Gastrointestinal Tract. J. Nutr. 2003, 133, 3255S–3261S. [Google Scholar] [CrossRef] [Green Version]
- Williamson, G.; Clifford, M.N. Role of the small intestine, colon and microbiota in determining the metabolic fate of polyphenols. Biochem. Pharm. 2017, 139, 24–39. [Google Scholar] [CrossRef]
- Kawabata, K.; Yoshioka, Y.; Terao, J. Role of Intestinal Microbiota in the Bioavailability and Physiological Functions of Dietary Polyphenols. Molecules 2019, 24, 370. [Google Scholar] [CrossRef] [Green Version]
- Mosele, J.I.; Macià, A.; Motilva, M.-J. Metabolic and Microbial Modulation of the Large Intestine Ecosystem by Non-Absorbed Diet Phenolic Compounds: A Review. Molecules 2015, 20, 17429–17468. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.H.; Dejea, C.M.; Edler, D.; Hoang, L.T.; Santidrian, A.F.; Felding, B.H.; Ivanisevic, J.; Cho, K.; Wick, E.C.; Hechenbleikner, E.M.; et al. Metabolism Links Bacterial Biofilms and Colon Carcinogenesis. Cell Metab. 2015. [Google Scholar] [CrossRef] [Green Version]
- Sonnenburg, J.L.; Backhed, F. Diet-microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef]
- Goodman, A.L.; Kallstrom, G.; Faith, J.J.; Reyes, A.; Moore, A.; Dantas, G.; Gordon, J.I. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc. Natl. Acad. Sci. USA 2011, 108, 6252–6257. [Google Scholar] [CrossRef] [Green Version]
- Wissenbach, D.K.; Oliphant, K.; Rolle-Kampczyk, U.; Yen, S.; Höke, H.; Baumann, S.; Haange, S.B.; Verdu, E.F.; Allen-Vercoe, E.; von Bergen, M. Optimization of metabolomics of defined in vitro gut microbial ecosystems. Int. J. Med. Microbiol. 2016, 306, 280–289. [Google Scholar] [CrossRef]
- Vanden Bussche, J.; Marzorati, M.; Laukens, D.; Vanhaecke, L. Validated High Resolution Mass Spectrometry-Based Approach for Metabolomic Fingerprinting of the Human Gut Phenotype. Anal. Chem. 2015. [Google Scholar] [CrossRef]
- Marzorati, M.; Vanhoecke, B.; De Ryck, T.; Sadaghian Sadabad, M.; Pinheiro, I.; Possemiers, S.; Van den Abbeele, P.; Derycke, L.; Bracke, M.; Pieters, J.; et al. The HMITM module: A new tool to study the Host-Microbiota Interaction in the human gastrointestinal tract in vitro. BMC Microbiol. 2014, 14, 133. [Google Scholar] [CrossRef] [Green Version]
- Macfarlane, G.T.; Macfarlane, S.; Gibson, G.R. Validation of a Three-Stage Compound Continuous Culture System for Investigating the Effect of Retention Time on the Ecology and Metabolism of Bacteria in the Human Colon. Microb. Ecol. 1998, 35, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.F.; Walton, G.E.; Jiang, L.; Plummer, S.; Garaiova, I.; Gibson, G.R. Comparative Analysis of Intestinal Tract Models. Annu. Rev. Food Sci. Technol. 2015, 6, 329–350. [Google Scholar] [CrossRef]
- Van de Wiele, T.; Van den Abbeele, P.; Ossieur, W.; Possemiers, S.; Marzorati, M. The Simulator of the Human Intestinal Microbial Ecosystem (SHIME®). In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Verhoeckx, K., Cotter, P., López-Expósito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 305–317. [Google Scholar] [CrossRef] [Green Version]
- Van de Wiele, T.R.; Oomen, A.G.; Wragg, J.; Cave, M.; Minekus, M.; Hack, A.; Cornelis, C.; Rompelberg, C.J.M.; De Zwart, L.L.; Klinck, B.; et al. Comparison of five in vitro digestion models to in vivo experimental results: Lead bioaccessibility in the human gastrointestinal tract. J. Environ. Sci. Health Part A 2007, 42, 1203–1211. [Google Scholar] [CrossRef]
- Boever, P.D.; Deplancke, B.; Verstraete, W. Fermentation by Gut Microbiota Cultured in a Simulator of the Human Intestinal Microbial Ecosystem Is Improved by Supplementing a Soygerm Powder. J. Nutr. 2000, 130, 2599–2606. [Google Scholar] [CrossRef]
- MÄKivuokko, H.A.; Saarinen, M.T.; Ouwehand, A.C.; Rautonen, N.E. Effects of Lactose on Colon Microbial Community Structure and Function in a Four-Stage Semi-Continuous Culture System. Biosci. Biotechnol. Biochem. 2006, 70, 2056–2063. [Google Scholar] [CrossRef]
- Daguet, D.; Pinheiro, I.; Verhelst, A.; Possemiers, S.; Marzorati, M. Arabinogalactan and fructooligosaccharides improve the gut barrier function in distinct areas of the colon in the Simulator of the Human Intestinal Microbial Ecosystem. J. Funct. Foods 2016, 20, 369–379. [Google Scholar] [CrossRef]
- Kuriyama, S.; Shimazu, T.; Ohmori, K.; Kikuchi, N.; Nakaya, N.; Nishino, Y.; Tsubono, Y.; Tsuji, I. Green tea consumption and mortality due to cardiovascular disease, cancer, and all causes in Japan: The Ohsaki study. JAMA 2006, 296, 1255–1265. [Google Scholar] [CrossRef]
- Krook, M.A.; Hagerman, A.E. Stability of Polyphenols Epigallocatechin Gallate and Pentagalloyl Glucose in a Simulated Digestive System. Food Res. Int. 2012, 49, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Herigstad, B.; Hamilton, M.; Heersink, J. How to optimize the drop plate method for enumerating bacteria. J. Microbiol. Methods 2001, 44, 121–129. [Google Scholar] [CrossRef]
- Zhao, D.; Shah, N.P. Tea and soybean extracts in combination with milk fermentation inhibit growth and enterocyte adherence of selected foodborne pathogens. Food Chem. 2015, 180, 306–316. [Google Scholar] [CrossRef]
- Liu, X.; Ser, Z.; Locasale, J.W. Development and quantitative evaluation of a high-resolution metabolomics technology. Anal. Chem. 2014, 86, 2175–2184. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Zhong, F.; Zhu, J. Evaluating metabolic response to light exposure in Lactobacillus species via targeted metabolic profiling. J. Microbiol. Methods 2017, 133, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Prabhu, S.; Meng, X.; Li, C.; Yang, C.S. An improved method for the determination of green and black tea polyphenols in biomatrices by high-performance liquid chromatography with coulometric array detection. Anal. Biochem. 2000, 279, 164–169. [Google Scholar] [CrossRef]
- Yang, K.; Duley, M.L.; Zhu, J. Metabolomics Study Reveals Enhanced Inhibition and Metabolic Dysregulation in Escherichia coli Induced by Lactobacillus acidophilus-Fermented Black Tea Extract. J. Agric. Food Chem. 2018, 66, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhong, F.; Bruno, R.S.; Ballard, K.D.; Zhang, J.; Zhu, J. Comparative Metabolomics Elucidates Postprandial Metabolic Modifications in Plasma of Obese Individuals with Metabolic Syndrome. J. Proteome Res. 2018, 17, 2850–2860. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, J. HPLC-MS/MS targeted metabolic profiling reveals distinct metabolic profiles from Staphylococcus aureus small-colony variants. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1060, 340–346. [Google Scholar] [CrossRef]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S. gplots: Various R programming tools for plotting data. R Package Version 2009, 2, 1. [Google Scholar]
- Chong, J.; Xia, J. MetaboAnalystR: An R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 2018, 34, 4313–4314. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Kemperman, R.A.; Gross, G.; Mondot, S.; Possemiers, S.; Marzorati, M.; Van de Wiele, T.; Doré, J.; Vaughan, E.E. Impact of polyphenols from black tea and red wine/grape juice on a gut model microbiome. Food Res. Int. 2013, 53, 659–669. [Google Scholar] [CrossRef]
- Hartung, T. Thoughts on limitations of animal models. Parkinsonism Relat. Disord. 2008, 14, S81–S83. [Google Scholar] [CrossRef]
- Van den Abbeele, P.; Grootaert, C.; Marzorati, M.; Possemiers, S.; Verstraete, W.; Gérard, P.; Rabot, S.; Bruneau, A.; El Aidy, S.; Derrien, M.; et al. Microbial Community Development in a Dynamic Gut Model Is Reproducible, Colon Region Specific, and Selective for Bacteroidetes and Clostridium Cluster IX. Appl. Environ. Microbiol. 2010, 76, 5237–5246. [Google Scholar] [CrossRef] [Green Version]
- Moreno, S.; Scheyer, T.; Romano, C.S.; Vojnov, A.A. Antioxidant and antimicrobial activities of rosemary extracts linked to their polyphenol composition. Free Radic. Res. 2006, 40, 223–231. [Google Scholar] [CrossRef]
- Middleton, E.; Kandaswami, C.; Theoharides, T.C. The effects of plant flavonoids on mammalian cells: Implications for inflammation, heart disease, and cancer. Pharmacol. Rev. 2000, 52, 673–751. [Google Scholar]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar] [CrossRef] [Green Version]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022. [Google Scholar] [CrossRef]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, T.; Derbyshire, E. Tea Compounds and the Gut Microbiome: Findings from Trials and Mechanistic Studies. Nutrients 2019, 11, 2364. [Google Scholar] [CrossRef] [Green Version]
- Jung, E.S.; Park, J.i.; Park, H.; Holzapfel, W.; Hwang, J.S.; Lee, C.H. Seven-day Green Tea Supplementation Revamps Gut Microbiome and Caecum/Skin Metabolome in Mice from Stress. Sci. Rep. 2019, 9, 18418. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Long, Y.; Ji, Z.; Gao, J.; Fu, T.; Yan, M.; Zhang, L.; Su, H.; Zhang, W.; Wen, X.; et al. Green Tea Liquid Consumption Alters the Human Intestinal and Oral Microbiome. Mol. Nutr. Food Res. 2018, 62, e1800178. [Google Scholar] [CrossRef] [Green Version]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-S.; Montana, V.; Jang, H.-J.; Parpura, V.; Kim, J.-A. Epigallocatechin-gallate (EGCG) stimulates autophagy in vascular endothelial cells: A potential role for reducing lipid accumulation. J. Biol. Chem. 2013, 288, 22693–22705. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.X.; Ritchie, S.R.; Rainey, P.B. Urocanate as a potential signaling molecule for bacterial recognition of eukaryotic hosts. Cell. Mol. Life Sci. 2014, 71, 541–547. [Google Scholar] [CrossRef]
- Chauhan, A.; Srivastva, N.; Bubber, P. Thiamine Deficiency Induced Dietary Disparity Promotes Oxidative Stress and Neurodegeneration. Indian J. Clin. Biochem. Ijcb 2018, 33, 422–428. [Google Scholar] [CrossRef]
- Ma, N.; Ma, X. Dietary Amino Acids and the Gut-Microbiome-Immune Axis: Physiological Metabolism and Therapeutic Prospects. Compr. Rev. Food Sci. Food Saf. 2019, 18, 221–242. [Google Scholar] [CrossRef] [Green Version]
- Keller, L.; Surette, M.G. Communication in bacteria: An ecological and evolutionary perspective. Nat. Rev. Microbiol. 2006, 4, 249–258. [Google Scholar] [CrossRef]







Sample Availability
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, M.; Yang, K.; Zhu, J. Monitoring the Diversity and Metabolic Shift of Gut Microbes during Green Tea Feeding in an In Vitro Human Colonic Model. Molecules 2020, 25, 5101. https://doi.org/10.3390/molecules25215101
Xu M, Yang K, Zhu J. Monitoring the Diversity and Metabolic Shift of Gut Microbes during Green Tea Feeding in an In Vitro Human Colonic Model. Molecules. 2020; 25(21):5101. https://doi.org/10.3390/molecules25215101
Chicago/Turabian StyleXu, Mengyang, Kundi Yang, and Jiangjiang Zhu. 2020. "Monitoring the Diversity and Metabolic Shift of Gut Microbes during Green Tea Feeding in an In Vitro Human Colonic Model" Molecules 25, no. 21: 5101. https://doi.org/10.3390/molecules25215101
APA StyleXu, M., Yang, K., & Zhu, J. (2020). Monitoring the Diversity and Metabolic Shift of Gut Microbes during Green Tea Feeding in an In Vitro Human Colonic Model. Molecules, 25(21), 5101. https://doi.org/10.3390/molecules25215101