Cinnamides Target Leishmania amazonensis Arginase Selectively

 ,
, 
 ,
, 
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Arginase Inhibition and Antileishmanial Activity
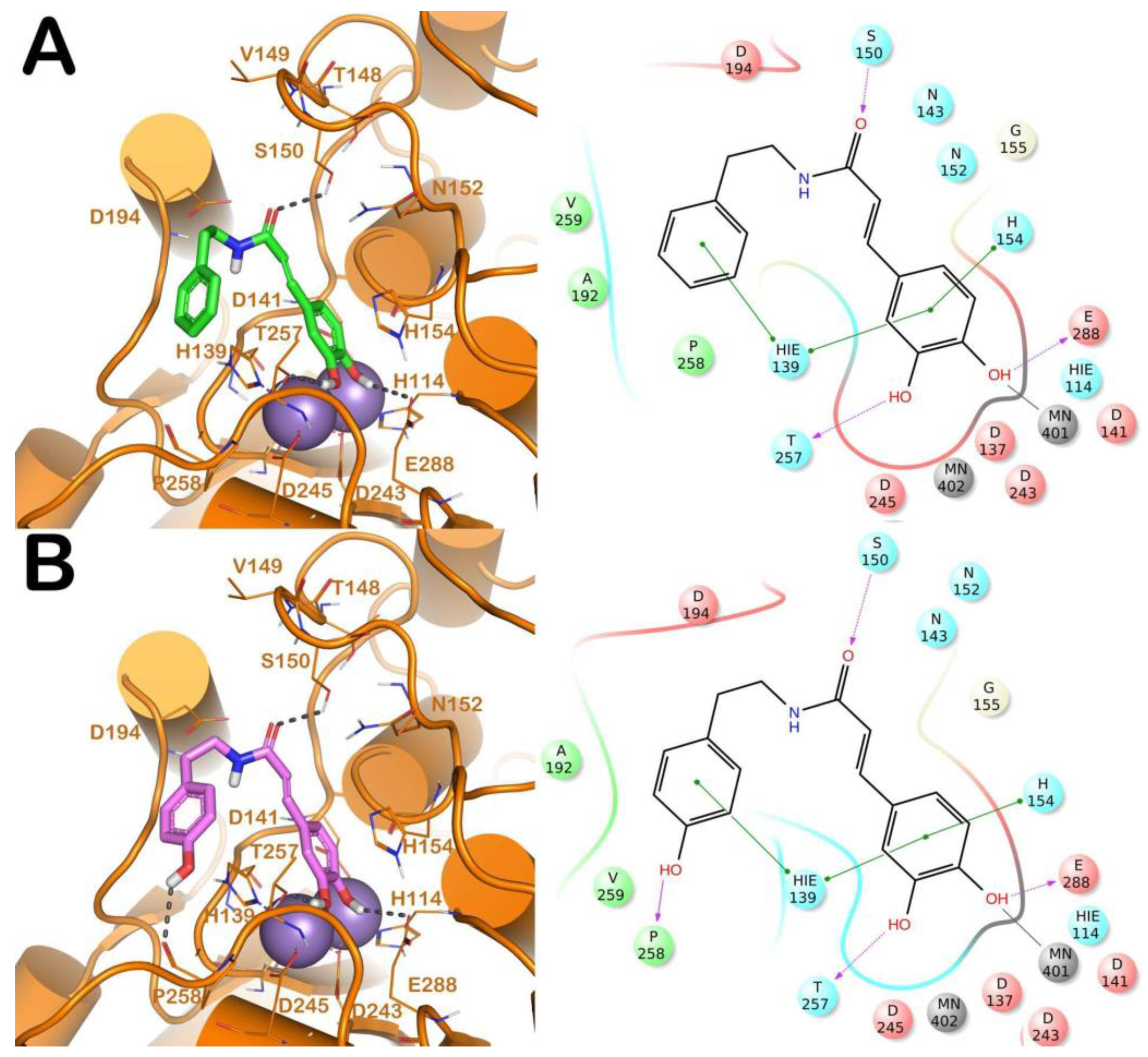
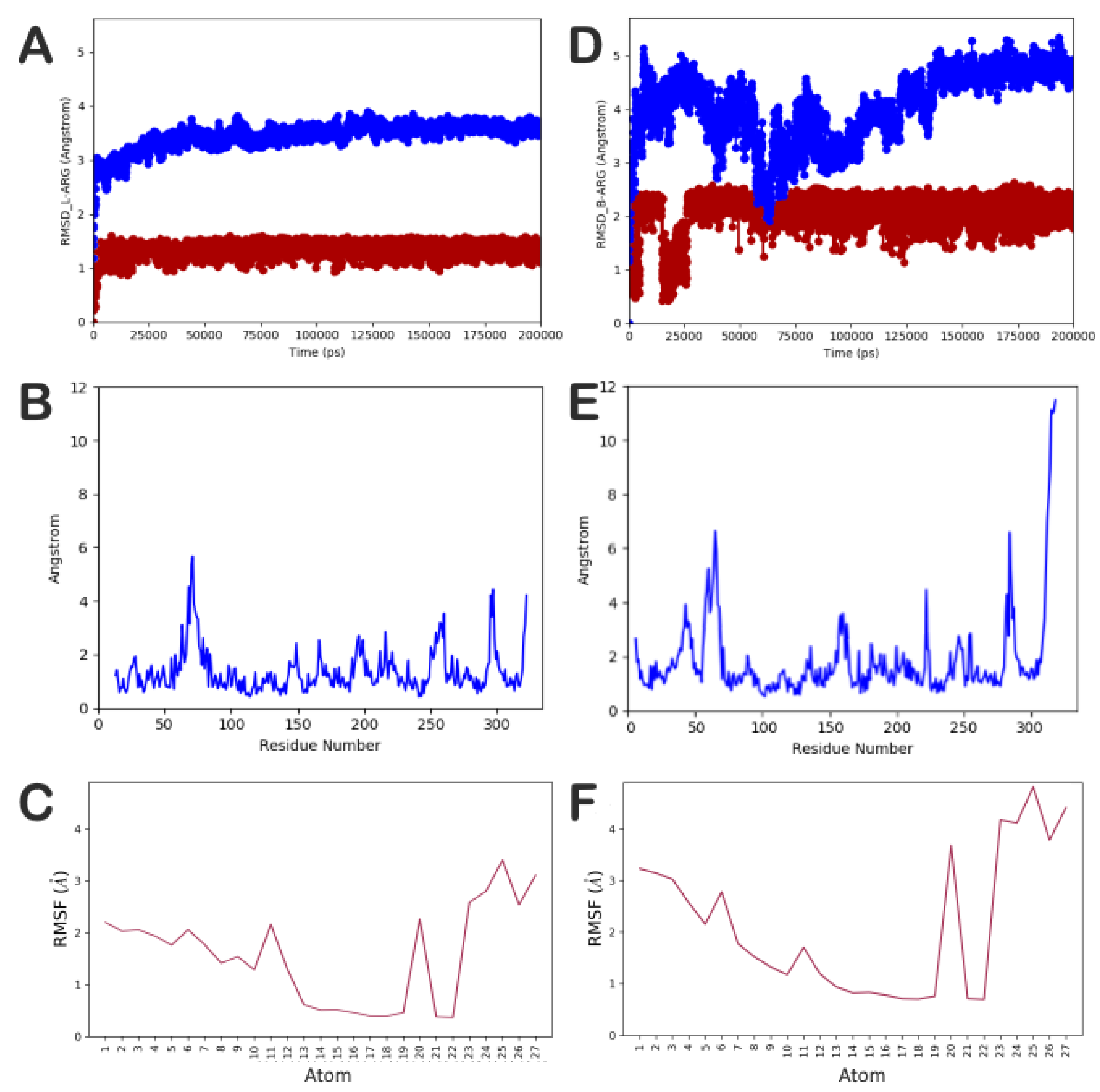
2.2. Computational Studies
Molecular Docking Studies, Homology Modeling, Binding Sites Analysis, and Molecular Dynamics Simulation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Expression and Purification of Arginase
4.3. Arginase Inhibition and Kinetics
4.4. Determination of the Constants Ki, Ki’ and the Mechanism of Inhibition
4.5. Promastigotes Culture
4.6. Computational Details
4.7. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Leishmaniasis. WHO. 2018. Available online: https://www.who.int/leishmaniasis/en/ (accessed on 25 January 2019).
- Tiwari, N.; Gedda, M.R.; Tiwari, V.K.; Singh, S.P.; Singh, R.K.; Tiwari, M.R.G.N. Limitations of Current Therapeutic Options, Possible Drug Targets and Scope of Natural Products in Control of Leishmaniasis. Mini Rev. Med. Chem. 2017, 18, 1. [Google Scholar] [CrossRef] [PubMed]
- Brito, N.C.; Rabello, A.; Cota, G. Efficacy of pentavalent antimoniate intralesional infiltration therapy for cutaneous leishmaniasis: A systematic review. PLoS ONE 2017, 12, e0184777. [Google Scholar] [CrossRef] [PubMed]
- Mosimann, V.; Neumayr, A.; Paris, D.H.; Blum, J. Liposomal amphotericin B treatment of Old World cutaneous and mucosal leishmaniasis: A literature review. Acta Trop. 2018, 182, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Christen, J.-R.; Bourreau, E.; Demar, M.; Lightburn, E.; Couppié, P.; Ginouvès, M.; Prévot, G.; Gangneux, J.-P.; Savini, H.; De Laval, F.; et al. Use of the intramuscular route to administer pentamidine isethionate in Leishmania guyanensis cutaneous leishmaniasis increases the risk of treatment failure. Travel Med. Infect. Dis. 2018, 24, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monge-Maillo, B.; López-Vélez, R. Miltefosine for Visceral and Cutaneous Leishmaniasis: Drug Characteristics and Evidence-Based Treatment Recommendations. Clin. Infect. Dis. 2015, 60, 1398–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.-C.; Barrett, M.P.; López-Vélez, R.; García-Hernández, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, e0006052. [Google Scholar] [CrossRef] [PubMed]
- Cipriano, P.; Miranda, A.C.; Antunes, I.; Mansinho, K. Leishmaniose Visceral em Doentes com Infeção VIH: O Desafio da Recaída e Falência Terapêutica. Acta Med. Port. 2017, 30, 443. [Google Scholar] [CrossRef] [Green Version]
- Vincendeau, P.; Gobert, A.P.; Daulouède, S.; Moynet, D.; Mossalayi, M.D. Arginases in parasitic diseases. Trends Parasitol. 2003, 19, 9–12. [Google Scholar] [CrossRef]
- Boitz, J.M.; Gilroy, C.A.; Olenyik, T.D.; Paradis, D.; Perdeh, J.; Dearman, K.; Davis, M.J.; Yates, P.A.; Li, Y.; Riscoe, M.K.; et al. Arginase Is Essential for Survival of Leishmania donovani Promastigotes but Not Intracellular Amastigotes. Infect. Immun. 2017, 85, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Fairlamb, A.H.; Cerami, A. Metabolism and Functions of Trypanothione in the Kinetoplastida. Annu. Rev. Microbiol. 1992, 46, 695–729. [Google Scholar] [CrossRef]
- Badirzadeh, A.; Etaheri, T.; Abedi-Astaneh, F.; Taslimi, Y.; Abdossamadi, Z.; Montakhab-Yeganeh, H.; Aghashahi, M.; Niyyati, M.; Rafati, S. Arginase activity ofLeishmaniaisolated from patients with cutaneous leishmaniasis. Parasite Immunol. 2017, 39, e12454. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.W.; Rodriguez, P.C.; Toque, H.A.; Narayanan, S.P. Arginase: A Multifaceted Enzyme Important in Health and Disease. Physiol. Rev. 2018, 98, 641–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montrieux, E.; Perera, W.H.; García, M.; Maes, L.; Cos, P.; Monzote, L. In vitro and in vivo activity of major constituents from Pluchea carolinensis against Leishmania amazonensis. Parasitol. Res. 2014, 113, 2925–2932. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, E.R.; Brogi, S.; Grillo, A.; Campiani, G.; Gemma, S.; Vieira, P.C.; Maquiaveli, C.D.C. Cinnamic acids derived compounds with antileishmanial activity target Leishmania amazonensis arginase. Chem. Biol. Drug Des. 2018, 93, 139–146. [Google Scholar] [CrossRef]
- Pham, T.-N.; Bordage, S.; Pudlo, M.; Demougeot, C.; Thai, K.-M.; Girard-Thernier, C. Cinnamide Derivatives as Mammalian Arginase Inhibitors: Synthesis, Biological Evaluation and Molecular Docking. Int. J. Mol. Sci. 2016, 17, 1656. [Google Scholar] [CrossRef]
- Maquiaveli, C.C.; Lucon-Júnior, J.F.; Brogi, S.; Campiani, G.; Gemma, S.; Vieira, P.C.; Silva, E.R. Verbascoside Inhibits Promastigote Growth and Arginase Activity of Leishmania amazonensis. J. Nat. Prod. 2016, 79, 1459–1463. [Google Scholar] [CrossRef]
- Da Silva, E.R.; Brogi, S.; Lucon-Júnior, J.F.; Campiani, G.; Gemma, S.; Maquiaveli, C.D.C. Dietary polyphenols rutin, taxifolin and quercetin related compounds target Leishmania amazonensis arginase. Food Funct. 2019, 10, 3172–3180. [Google Scholar] [CrossRef]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nat. Cell Biol. 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Dos Reis, M.B.G.; Manjolin, L.C.; Maquiaveli, C.D.C.; Santos-Filho, O.A.; Da Silva, E.R. Inhibition of Leishmania (Leishmania) amazonensis and Rat Arginases by Green Tea EGCG, (+)-Catechin and (−)-Epicatechin: A Comparative Structural Analysis of Enzyme-Inhibitor Interactions. PLoS ONE 2013, 8, e78387. [Google Scholar] [CrossRef]
- Da Silva, E.R.; Maquiaveli, C.D.C.; Magalhães, P.P. The leishmanicidal flavonols quercetin and quercitrin target Leishmania (Leishmania) amazonensis arginase. Exp. Parasitol. 2012, 130, 183–188. [Google Scholar] [CrossRef]
- Manjolin, L.C.; Dos Reis, M.B.G.; Maquiaveli, C.D.C.; Santos-Filho, O.A.; Da Silva, E.R. Dietary flavonoids fisetin, luteolin and their derived compounds inhibit arginase, a central enzyme in Leishmania (Leishmania) amazonensis infection. Food Chem. 2013, 141, 2253–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordage, S.; Pham, T.-N.; Zedet, A.; Gugglielmetti, A.-S.; Nappey, M.; Demougeot, C.; Girard-Thernier, C. Investigation of Mammal Arginase Inhibitory Properties of Natural Ubiquitous Polyphenols by Using an Optimized Colorimetric Microplate Assay. Planta Med. 2016, 83, 647–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, E.R.; Castilho, T.M.; Pioker, F.C.; Silva, C.H.T.D.P.D.; Winter, L.M.F. Genomic organisation and transcription characterisation of the gene encoding Leishmania (Leishmania) amazonensis arginase and its protein structure prediction. Int. J. Parasitol. 2002, 32, 727–737. [Google Scholar] [CrossRef]
- D’Antonio, E.L.; Ullman, B.; Roberts, S.C.; Dixit, U.G.; Wilson, M.E.; Hai, Y.; Christianson, D.W. Crystal structure of arginase from Leishmania mexicana and implications for the inhibition of polyamine biosynthesis in parasitic infections. Arch. Biochem. Biophys. 2013, 535, 163–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, E.; Roberts, S.C.; Ullman, B. Inhibition profile of Leishmania mexicana arginase reveals differences with human arginase I. Int. J. Parasitol. 2011, 41, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Feitosa, L.M.; Da Silva, E.R.; Hoelz, L.V.; Souza, D.L.; Come, J.A.; Cardoso-Santos, C.; Batista, M.M.; Soeiro, M.D.N.C.; Boechat, N.; Pinheiro, L.C. New pyrazolopyrimidine derivatives as Leishmania amazonensis arginase inhibitors. Bioorg. Med. Chem. 2019, 27, 3061–3069. [Google Scholar] [CrossRef]
- Da Silva, E.R.; Boechat, N.; Pinheiro, L.C.; Bastos, M.M.; Costa, C.C.P.; Bartholomeu, J.C.; Da Costa, T.H. Novel Selective Inhibitor of Leishmania (Leishmania) amazonensis Arginase. Chem. Biol. Drug Des. 2015, 86, 969–978. [Google Scholar] [CrossRef]
- De Lima, E.C.; Castelo-Branco, F.S.; Maquiaveli, C.C.; Farias, A.B.; Rennó, M.N.; Boechat, N.; Da Silva, E.R. Phenylhydrazides as inhibitors of Leishmania amazonensis arginase and antileishmanial activity. Bioorg. Med. Chem. 2019, 27, 3853–3859. [Google Scholar] [CrossRef]
- Da Silva, E.R.; Laranjeira-Silva, M.F.; Fischer, H.; Mortara, R.A.; Mayer, M.G.; Framesqui, K.; Silber, A.M.; Winter, L.M.F. Biochemical and biophysical properties of a highly active recombinant arginase from Leishmania (Leishmania) amazonensis and subcellular localization of native enzyme. Mol. Biochem. Parasitol. 2008, 159, 104–111. [Google Scholar] [CrossRef]
- Fawcett, J.K.; Scott, J.E. A rapid and precise method for the determination of urea. J. Clin. Pathol. 1960, 13, 156–159. [Google Scholar] [CrossRef] [Green Version]
- Cornish-Bowden, A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors (Short Communication). Biochem. J. 1974, 137, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Gasser, A.; Brogi, S.; Urayama, K.; Nishi, T.; Kurose, H.; Tafi, A.; Ribeiro, N.; Désaubry, L.; Nebigil, C.G. Discovery and Cardioprotective Effects of the First Non-Peptide Agonists of the G Protein-Coupled Prokineticin Receptor-1. PLoS ONE 2015, 10, e0121027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaccagnini, L.; Brogi, S.; Brindisi, M.; Gemma, S.; Chemi, G.; Legname, G.; Campiani, G.; Butini, S. Identification of novel fluorescent probes preventing PrP Sc replication in prion diseases. Eur. J. Med. Chem. 2017, 127, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides†. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Chemi, G.; Gemma, S.; Campiani, G.; Brogi, S.; Butini, S.; Brindisi, M. Computational Tool for Fast in silico Evaluation of hERG K+ Channel Affinity. Front. Chem. 2017, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Sirous, H.; Chemi, G.; Campiani, G.; Brogi, S. An integrated in silico screening strategy for identifying promising disruptors of p53-MDM2 interaction. Comput. Biol. Chem. 2019, 83, 107105. [Google Scholar] [CrossRef]
- Brogi, S.; Giovani, S.; Brindisi, M.; Gemma, S.; Novellino, E.; Campiani, G.; Blackman, M.J.; Butini, S. In silico study of subtilisin-like protease 1 (SUB1) from different Plasmodium species in complex with peptidyl-difluorostatones and characterization of potent pan-SUB1 inhibitors. J. Mol. Graph. Model. 2016, 64, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Sirous, H.; Fassihi, A.; Brogi, S.; Campiani, G.; Christ, F.; Debyser, Z.; Gemma, S.; Butini, S.; Chemi, G.; Grillo, A.; et al. Synthesis, Molecular Modelling and Biological Studies of 3-hydroxypyrane- 4-one and 3-hydroxy-pyridine-4-one Derivatives as HIV-1 Integrase Inhibitors. Med. Chem. 2019, 15, 755–770. [Google Scholar] [CrossRef]
- Caruso, U.; Ilies, M.; Thorn, K.J.; Christianson, D.W. Inhibition of human arginase I by substrate and product analogues. Arch. Biochem. Biophys. 2010, 496, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Sirous, H.; Chemi, G.; Gemma, S.; Butini, S.; Debyser, Z.; Christ, F.; Saghaie, L.; Brogi, S.; Fassihi, A.; Campiani, G.; et al. Identification of Novel 3-Hydroxy-pyran-4-One Derivatives as Potent HIV-1 Integrase Inhibitors Using in silico Structure-Based Combinatorial Library Design Approach. Front. Chem. 2019, 7, 574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolino, M.; Brindisi, M.; Vallone, A.; Butini, S.; Campiani, G.; Nannicini, C.; Giuliani, G.; Anzini, M.; Lamponi, S.; Giorgi, G.; et al. Development of Potent Inhibitors of the Mycobacterium tuberculosis Virulence Factor Zmp1 and Evaluation of Their Effect on Mycobacterial Survival inside Macrophages. ChemMedChem 2018, 13, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Nickolls, J.; Buck, I.; Garland, M.; Skadron, K. Scalable Parallel Programming with CUDA. Queue 2008, 6, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Brindisi, M.; Ulivieri, C.; Alfano, G.; Gemma, S.; Balaguer, F.D.A.; Khan, T.; Grillo, A.; Brindisi, M.; Menchon, G.; Prota, A.E.; et al. Structure-activity relationships, biological evaluation and structural studies of novel pyrrolonaphthoxazepines as antitumor agents. Eur. J. Med. Chem. 2019, 162, 290–320. [Google Scholar] [CrossRef]
- Humphreys, D.D.; Friesner, R.A.; Berne, B.J. A Multiple-Time-Step Molecular Dynamics Algorithm for Macromolecules. J. Phys. Chem. 1994, 98, 6885–6892. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | IC50 (µM) | Emax (%) | Ki (µM) | Kis (µM) | Mode of Inhibition |
|---|---|---|---|---|---|---|
| CAPA |  | 6.9 ± 0.7 | 79 ± 1 | 3.9 ±1.0 | - | Competitive |
| 6 |  | 5.6 ± 0.4 | 82 ± 2 | 4.4 ± 1.1 | - | Competitive |
| 11 |  | 1.8 ± 0.1 | 81 ± 2 | - | 2.5 ± 0.2 | Uncompetitive |
| 12 |  | 6.5 ± 1.4 | 75 ± 2 | 9.6 ± 0.7 | 9.6 ± 0.7 | Noncompetitive |
| 13 |  | 2.5 ± 0.2 | 82 ± 1 | 1.6 ± 0.2 | - | Competitive |
| 14 |  | 2.4 ± 0.9 | 81 ± 2 | 1.2 ± 0.3 | - | Competitive |
| 15 |  | 1.3 ± 01 | 80 ± 1 | 1.4 ± 0.2 | 5 ± 1 | Mixed |
| 17 |  | 18 ± 3 | 68 ± 2 | 11.4 ± 0.7 | - | Competitive |
| 18 |  | 4 ± 1 | 89 ± 3 | 3.9 ± 0.4 | 27 ± 8 | Mixed |
| 19 |  | 9 ± 1 | 90 ± 3 | 6.6 ± 0.3 | 28 ± 4 | Mixed |
| Compound | L. amazonensis | Bovine a | Selective Index |
|---|---|---|---|
| CAPA | 6.9 ± 0.7 | 6.9 ± 1.3 | 1 |
| 6 | 5.6 ± 0.4 | 22.1 ± 1.6 | 4 |
| 11 | 1.8 ± 0.1 | 114.9 ± 1.3 | 64 |
| 12 | 6.5 ± 1.4 | 198.7 ± 1.4 | 31 |
| 13 | 2.5 ± 0.2 | 170.4 ± 1.7 | 68 |
| 14 | 2.4 ± 0.9 | 193.6 ± 1.4 | 81 |
| 15 | 1.3 ± 0.1 | 39.3 ± 1.4 | 30 |
| 17 | 18 ± 3 | 175.3 ± 1.5 | 10 |
| 18 | 4 ± 1 | 41.9 ± 1.3 | 10 |
| 19 | 9 ± 1 | 37.0 ± 1.3 | 4 |
| Caffeic acid | 1.5 ± 0.3 b | 86.7 ± 8 | 58 |
| Chlorogenic acid | 8.3 ± 0.2 b | 10.6 ± 1.6 | 1.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva, E.R.; Come, J.A.A.d.S.S.; Brogi, S.; Calderone, V.; Chemi, G.; Campiani, G.; Oliveira, T.M.F.d.S.; Pham, T.-N.; Pudlo, M.; Girard, C.; et al. Cinnamides Target Leishmania amazonensis Arginase Selectively. Molecules 2020, 25, 5271. https://doi.org/10.3390/molecules25225271
da Silva ER, Come JAAdSS, Brogi S, Calderone V, Chemi G, Campiani G, Oliveira TMFdS, Pham T-N, Pudlo M, Girard C, et al. Cinnamides Target Leishmania amazonensis Arginase Selectively. Molecules. 2020; 25(22):5271. https://doi.org/10.3390/molecules25225271
Chicago/Turabian Styleda Silva, Edson Roberto, Júlio Abel Alfredo dos Santos Simone Come, Simone Brogi, Vincenzo Calderone, Giulia Chemi, Giuseppe Campiani, Trícia Maria Ferrreira de Sousa Oliveira, Thanh-Nhat Pham, Marc Pudlo, Corine Girard, and et al. 2020. "Cinnamides Target Leishmania amazonensis Arginase Selectively" Molecules 25, no. 22: 5271. https://doi.org/10.3390/molecules25225271
APA Styleda Silva, E. R., Come, J. A. A. d. S. S., Brogi, S., Calderone, V., Chemi, G., Campiani, G., Oliveira, T. M. F. d. S., Pham, T. -N., Pudlo, M., Girard, C., & Maquiaveli, C. d. C. (2020). Cinnamides Target Leishmania amazonensis Arginase Selectively. Molecules, 25(22), 5271. https://doi.org/10.3390/molecules25225271