
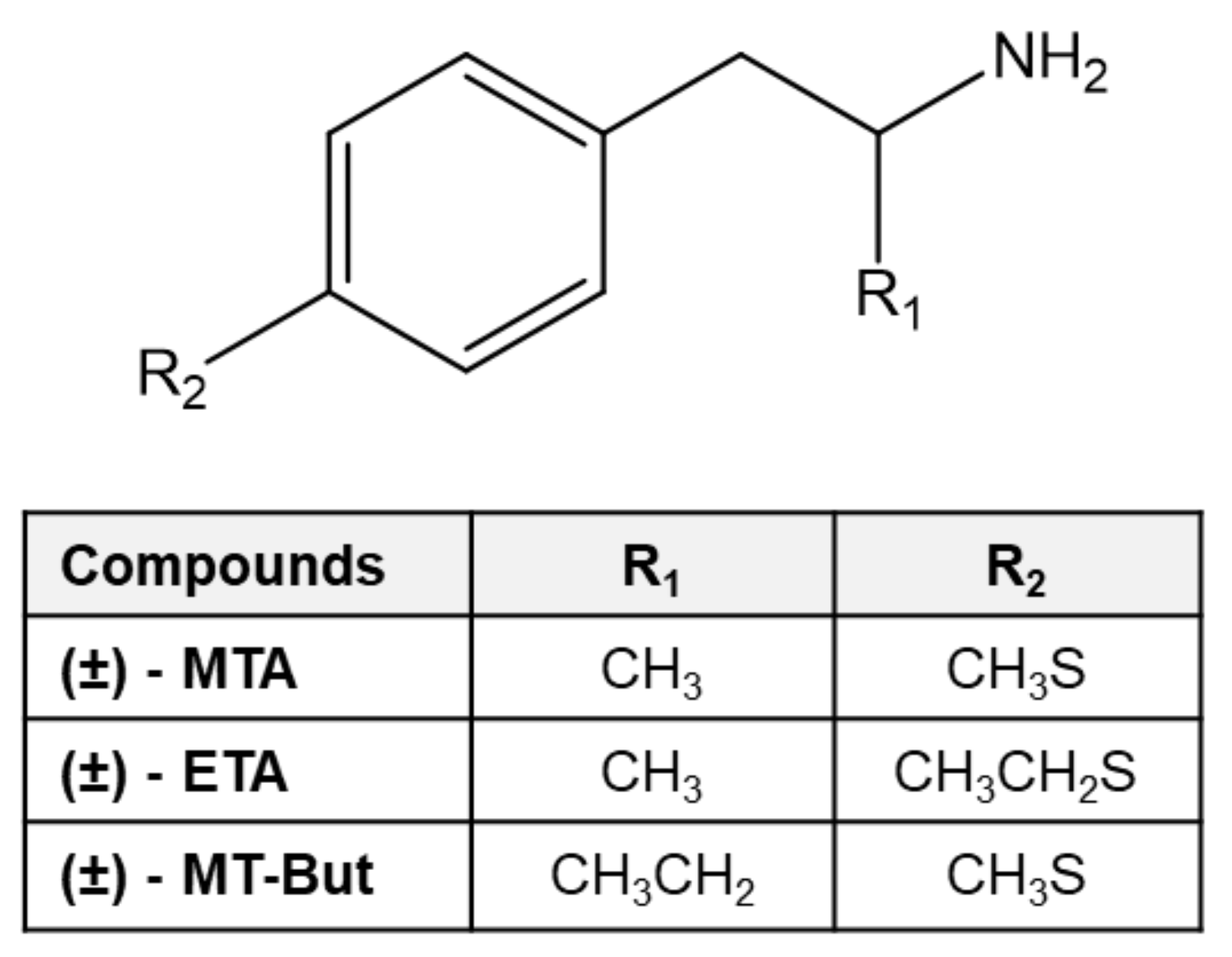
Pharmacological Characterization of 4-Methylthioamphetamine Derivatives

 , and
, and 
Abstract
:1. Introduction
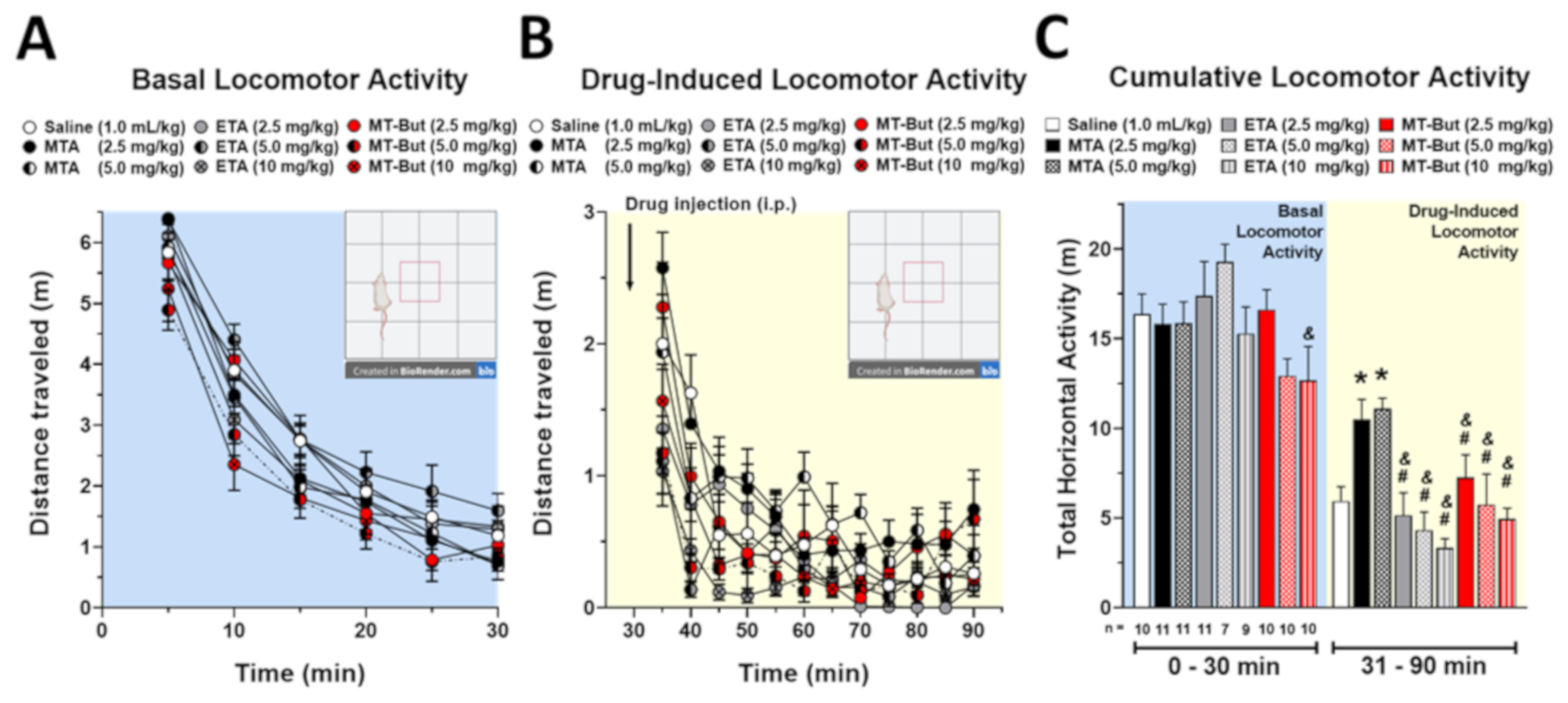
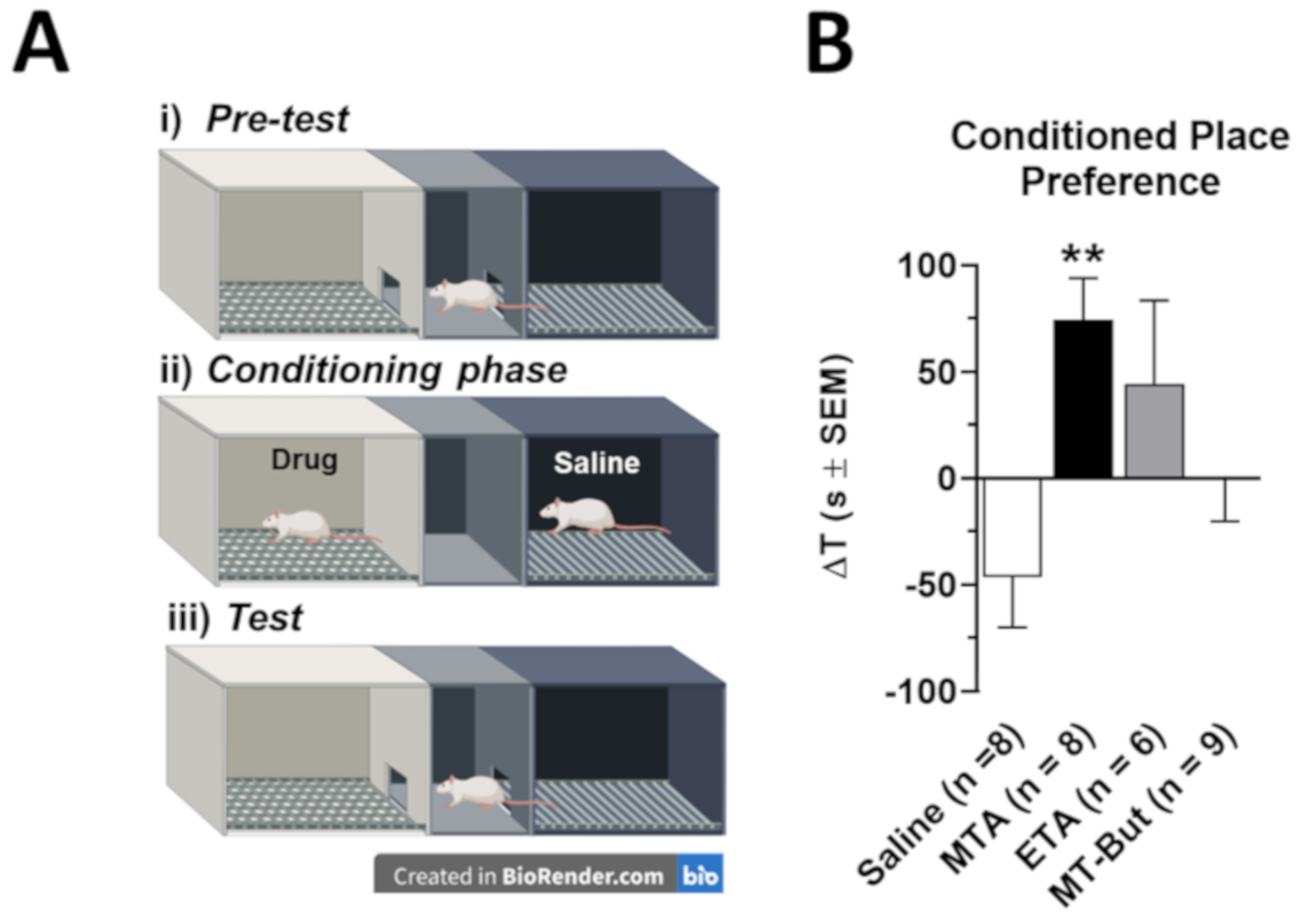
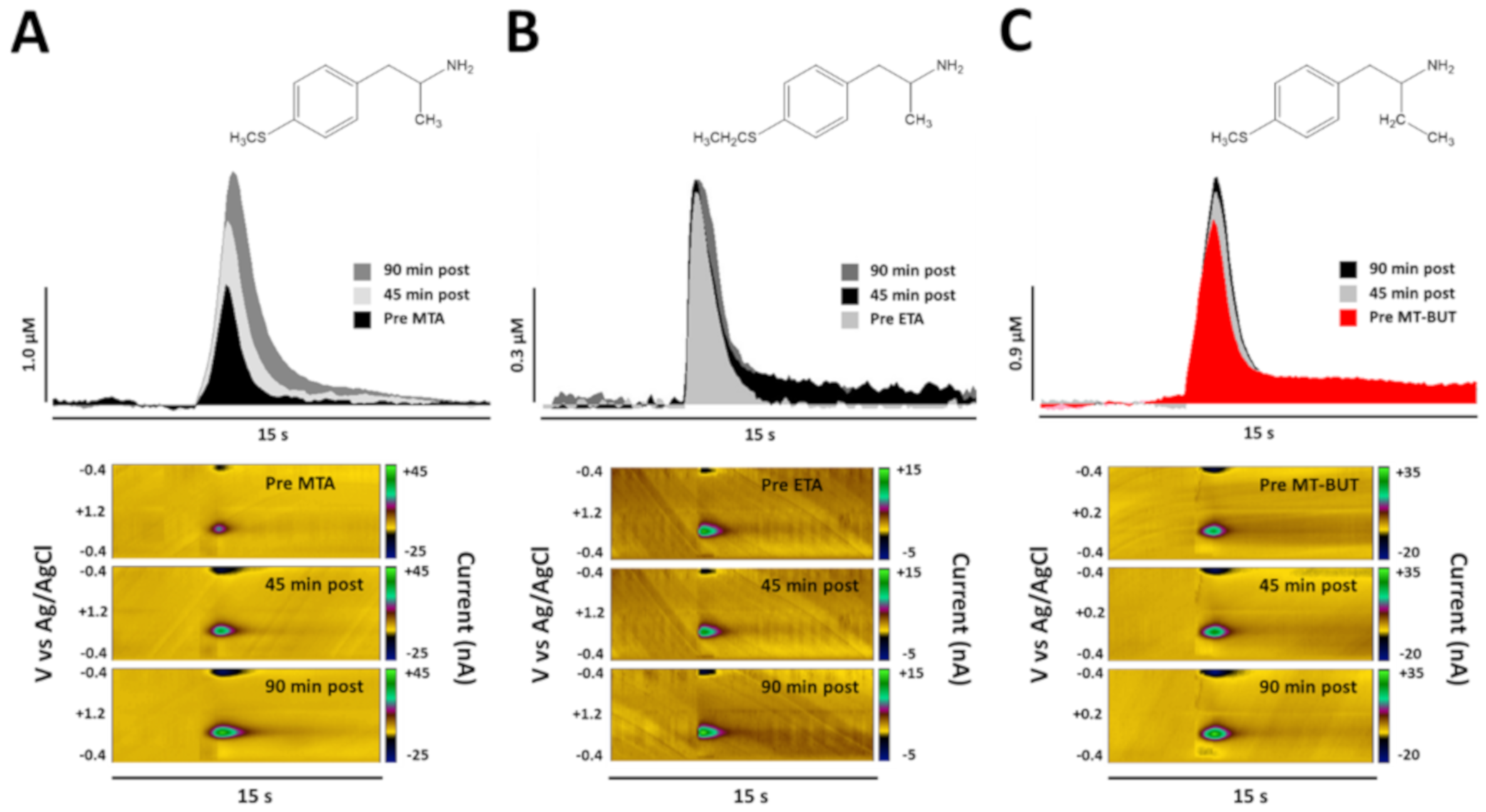
2. Results
2.1. Behavioral Data in Locomotor Activity and Conditioned Place Preference Tests
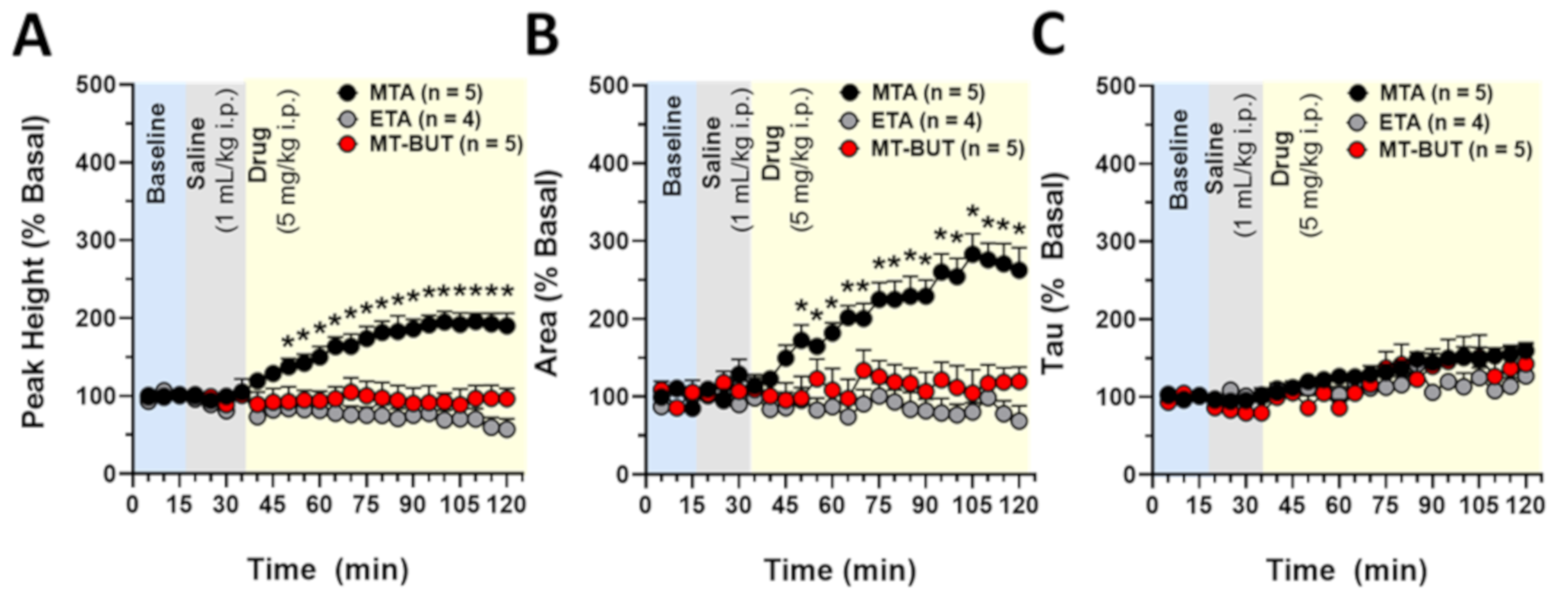
2.2. Neurochemical Data of Tissue Content and Release of DA
2.3. Cardiovascular Safety Profile
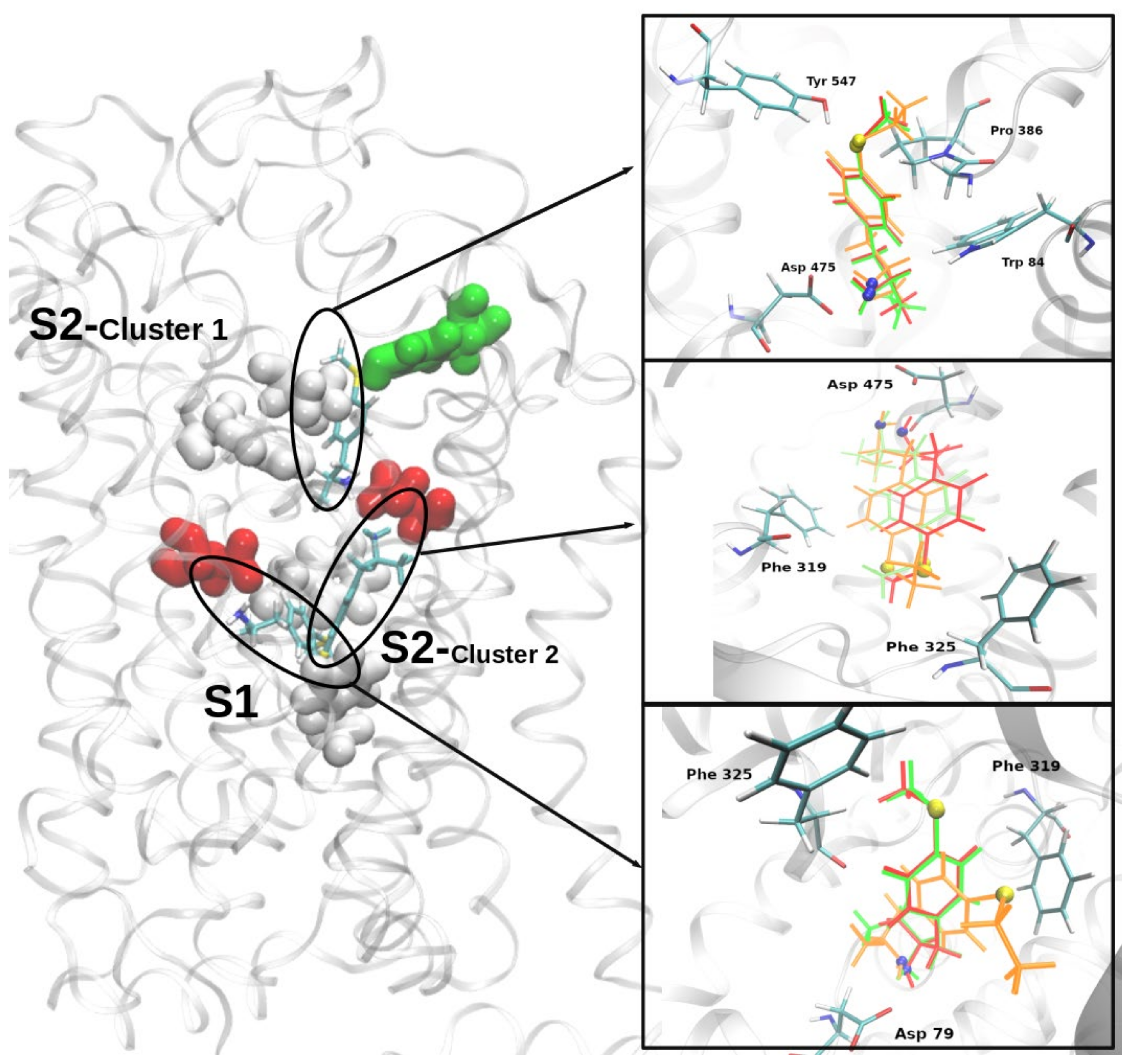
2.4. Molecular Docking of MTA, ETA, and MT-But on Rat (r)DAT Model
3. Discussion
3.1. Neurochemical Effects of MTA Derivatives
3.2. Behavioral Effects of MTA Derivatives
3.3. In Vivo Safety Profile of MTA Derivatives
3.4. In Silico Pharmacology of MTA, ETA, and MT-But on rDAT Model
4. Materials and Methods
4.1. Animals
4.2. Reagents
4.3. Behavioral Studies
4.3.1. Locomotor Activity
4.3.2. Conditional Preference Place (CPP)
4.4. Neurochemical Studies
4.4.1. NAcc DOPAC and DA Content Using HPLC Coupled to Electrochemical Detection
4.4.2. DA and DOPAC Quantification
4.4.3. In Vivo Fast Scan Cyclic Voltammetry to Measure DA Extracellular Levels in Dorsal Striatum
4.5. In Vivo Assessment of Blood Pressure, Heart Rate, and Body Temperature
4.6. Homology Modeling and Molecular Simulation
4.7. Molecular Docking
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fleckenstein, A.E.; Volz, T.J.; Riddle, E.L.; Gibb, J.W.; Hanson, G.R. New insights into the mechanism of action of amphetamines. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 681–698. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Sonders, M.S.; Poulsen, N.W.; Galli, A. Mechanisms of neurotransmitter release by amphetamines: A review. Prog. Neurobiol. 2005, 75, 406–433. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Maeno, Y.; Kato, H.; Seko-Nakamura, Y.; Monma-Ohtaki, J.; Ishiba, A.; Nagao, M.; Aoki, Y. 5-hydroxytryptamine- and dopamine-releasing effects of ring-substituted amphetamines on rat brain: A comparative study using in vivo microdialysis. Eur. Neuropsychopharmacol. 2014, 24, 1362–1370. [Google Scholar] [CrossRef]
- Scorza, M.C.; Carrau, C.; Silveira, R.; Zapata-Torres, G.; Cassels, B.K.; Reyes-Parada, M. Monoamine oxidase inhibitory properties of some methoxylated and alkylthio amphetamine derivatives: Structure-activity relationships. Biochem. Pharmacol. 1997, 54, 1361–1369. [Google Scholar] [CrossRef]
- Reyes-Parada, M.; Iturriaga-Vasquez, P.; Cassels, B.K. Amphetamine derivatives as monoamine oxidase inhibitors. Front. Pharmacol. 2020, 10, 1590. [Google Scholar] [CrossRef]
- Janardhanan, R.; Kannan, A. Methamphetamine cardiotoxicity: Unique presentation with multiple bi-ventricular thrombi. Am. J. Med. 2016, 129, e3–e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, D.W. Lisdexamfetamine dimesylate (vyvanse), a prodrug stimulant for attention-deficit/hyperactivity disorder. Pharm. Ther. 2010, 35, 273–287. [Google Scholar]
- Hilbert, A. Binge-eating disorder. Psychiatr. Clin. North Am. 2019, 42, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Mithoefer, M.C.; Wagner, M.T.; Mithoefer, A.T.; Jerome, L.; Martin, S.F.; Yazar-Klosinski, B.; Michel, Y.; Brewerton, T.D.; Doblin, R. Durability of improvement in post-traumatic stress disorder symptoms and absence of harmful effects or drug dependency after 3,4-methylenedioxymethamphetamine-assisted psychotherapy: A prospective long-term follow-up study. J. Psychopharmacol. 2013, 27, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Oehen, P.; Traber, R.; Widmer, V.; Schnyder, U. A randomized, controlled pilot study of MDMA (+/− 3,4-Methylenedioxymethamphetamine)-assisted psychotherapy for treatment of resistant, chronic post-traumatic stress disorder (PTSD). J. Psychopharmacol. 2013, 27, 40–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winstock, A.R.; Wolff, K.; Ramsey, J. 4-MTA: A new synthetic drug on the dance scene. Drug Alcohol Depend. 2002, 67, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Marona-Lewicka, D.; Nichols, D.E. p-methylthioamphetamine is a potent new non-neurotoxic serotonin-releasing agent. Eur. J. Pharmacol. 1992, 229, 31–38. [Google Scholar] [CrossRef]
- Gobbi, M.; Moia, M.; Pirona, L.; Ceglia, I.; Reyes-Parada, M.; Scorza, C.; Mennini, T. p-Methylthioamphetamine and 1-(m-chlorophenyl)piperazine, two non-neurotoxic 5-HT releasers in vivo, differ from neurotoxic amphetamine derivatives in their mode of action at 5-HT nerve endings in vitro. J. Neurochem. 2002, 82, 1435–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotomayor-Zarate, R.; Quiroz, G.; Araya, K.A.; Abarca, J.; Ibanez, M.R.; Montecinos, A.; Guajardo, C.; Nunez, G.; Fierro, A.; Moya, P.R.; et al. 4-Methylthioamphetamine increases dopamine in the rat striatum and has rewarding effects in vivo. Basic Clin. Pharmacol. Toxicol. 2012, 111, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Murakami, I.; Stall, S.; Levy, A.D.; Brownfield, M.S.; Nichols, D.E.; Van de Kar, L.D. Neuroendocrine pharmacology of three serotonin releasers: 1-(1,3-benzodioxol-5-yl)-2-(methylamino)butane (MBDB), 5-methoxy-6-methyl-2-aminoindan (MMAi) and p-methylthioamphetamine (MTA). J. Pharmacol. Exp. Ther. 1996, 279, 1261–1267. [Google Scholar]
- Scorza, C.; Silveira, R.; Nichols, D.E.; Reyes-Parada, M. Effects of 5-HT-releasing agents on the extracellullar hippocampal 5-HT of rats. Implications for the development of novel antidepressants with a short onset of action. Neuropharmacology 1999, 38, 1055–1061. [Google Scholar] [CrossRef]
- Sotomayor-Zarate, R.; Jara, P.; Araos, P.; Vinet, R.; Quiroz, G.; Renard, G.M.; Espinosa, P.; Hurtado-Guzman, C.; Moya, P.R.; Iturriaga-Vasquez, P.; et al. Improving amphetamine therapeutic selectivity: N,N-dimethyl-MTA has dopaminergic effects and does not produce aortic contraction. Basic Clin. Pharmacol. Toxicol. 2014, 114, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Gobbi, M.; Funicello, M.; Gerstbrein, K.; Holy, M.; Moya, P.R.; Sotomayor, R.; Forray, M.I.; Gysling, K.; Paluzzi, S.; Bonanno, G.; et al. N,N-dimethyl-thioamphetamine and methyl-thioamphetamine, two non-neurotoxic substrates of 5-HT transporters, have scant in vitro efficacy for the induction of transporter-mediated 5-HT release and currents. J. Neurochem. 2008, 105, 1770–1780. [Google Scholar] [CrossRef] [Green Version]
- Hurtado-Guzman, C.; Fierro, A.; Iturriaga-Vasquez, P.; Sepulveda-Boza, S.; Cassels, B.K.; Reyes-Parada, M. Monoamine oxidase inhibitory properties of optical isomers and N-substituted derivatives of 4-methylthioamphetamine. J. Enzym. Inhib. Med. Chem. 2003, 18, 339–347. [Google Scholar] [CrossRef]
- Di Chiara, G.; Imperato, A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. USA 1988, 85, 5274–5278. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.H.; Penmatsa, A.; Gouaux, E. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 2015, 521, 322–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, J.; Ladefoged, L.K.; Kristensen, T.N.; Munro, L.; Grouleff, J.; Stuhr-Hansen, N.; Kristensen, A.S.; Schiott, B.; Stromgaard, K. Interrogating the molecular basis for substrate recognition in serotonin and dopamine transporters with high-affinity substrate-based bivalent ligands. ACS Chem. Neurosci. 2016, 7, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Reith, M.E.A.; Gnegy, M.E. Molecular mechanisms of amphetamines. Handb. Exp. Pharmacol. 2020, 258, 265–297. [Google Scholar] [PubMed]
- Nichols, D.E. Medicinal chemistry and structure-activity relationships. In Amphetamine and Its Analogues: Psychopharmacology, Toxicology, and Abuse; Academic Press: San Diego, CA, USA, 1994; pp. 3–41. [Google Scholar]
- Hegadoren, K.M.; Greenshaw, A.J.; Baker, G.B.; Martin-Iverson, M.T.; Lodge, B.; Soin, S. 4-Ethoxyamphetamine: Effects on intracranial self-stimulation and in vitro uptake and release of 3H-dopamine and 3H-serotonin in the brains of rats. J. Psychiatry Neurosci. 1994, 19, 57–62. [Google Scholar]
- Nunez-Vivanco, G.; Fierro, A.; Moya, P.; Iturriaga-Vasquez, P.; Reyes-Parada, M. 3D similarities between the binding sites of monoaminergic target proteins. PLoS ONE 2018, 13, e0200637. [Google Scholar] [CrossRef] [PubMed]
- Golembiowska, K.; Jurczak, A.; Kaminska, K.; Noworyta-Sokolowska, K.; Gorska, A. Effect of some psychoactive drugs used as ‘legal highs’ on brain neurotransmitters. Neurotox. Res. 2016, 29, 394–407. [Google Scholar] [CrossRef] [Green Version]
- Zawilska, J.B.; Wojcieszak, J. Designer cathinones—An emerging class of novel recreational drugs. Forensic Sci. Int. 2013, 231, 42–53. [Google Scholar] [CrossRef]
- Morikawa, H.; Paladini, C.A. Dynamic regulation of midbrain dopamine neuron activity: Intrinsic, synaptic, and plasticity mechanisms. Neuroscience 2011, 198, 95–111. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, C.; Leitz, M.; Oberdorf-Maass, S.; Lohse, M.; Klotz, K.N. Comparative pharmacology of human β-adrenergic receptor subtypes—characterization of stably transfected receptors in CHO cells. Naunyn Schmiedeberg Arch. Pharmacol. 2004, 369, 151–159. [Google Scholar] [CrossRef]
- Bicego, K.C.; Barros, R.C.; Branco, L.G. Physiology of temperature regulation: Comparative aspects. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2007, 147, 616–639. [Google Scholar] [CrossRef]
- Rodríguez-Cuenca, S.; Pujol, E.; Justo, R.; Frontera, M.; Oliver, J.; Gianotti, M.; Roca, P. Sex-dependent thermogenesis. Differences in mitochondrial morphology and function, and adrenergic response in bat. J. Biol. Chem. 2002, 277, 42958–42963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coman, O.A.; Paunescu, H.; Ghita, I.; Coman, L.; Badararu, A.; Fulga, I. Beta 3 adrenergic receptors: Molecular, histological, functional and pharmacological approaches. Rom. J. Morphol. Embryol. 2009, 50, 169–179. [Google Scholar] [PubMed]
- Paulo, E.; Wu, D.; Wang, Y.; Zhang, Y.; Wu, Y.; Swaney, D.L.; Soucheray, M.; Jimenez-Morales, D.; Chawla, A.; Krogan, N.J. Sympathetic inputs regulate adaptive thermogenesis in brown adipose tissue through cAMP-Salt inducible kinase axis. Sci. Rep. 2018, 8, 11001. [Google Scholar] [CrossRef] [PubMed]
- Volpi-Abadie, J.; Kaye, A.M.; Kaye, A.D. Serotonin syndrome. Ochsner J. 2013, 13, 533–540. [Google Scholar]
- Quinn, S.T.; Guiry, P.J.; Schwab, T.; Keenan, A.K.; McBean, G.J. Blockade of noradrenaline transport abolishes 4-methylthioamphetamine-induced contraction of the rat aorta in vitro. Auton Autacoid Pharmacol. 2006, 26, 335–344. [Google Scholar] [CrossRef]
- Xu, L.; Chen, L.Y. Identification of a new allosteric binding site for cocaine in dopamine transporter. J. Chem. Inf. Model 2020, 60, 3958–3968. [Google Scholar] [CrossRef]
- Cruz, G.; Riquelme, R.; Espinosa, P.; Jara, P.; Dagnino-Subiabre, A.; Renard, G.M.; Sotomayor-Zarate, R. Neonatal exposure to estradiol valerate increases dopamine content in nigrostriatal pathway during adulthood in the rat. Horm. Metab. Res. Horm. Stoffwechs. Horm. Metab. 2014, 46, 322–327. [Google Scholar] [CrossRef] [Green Version]
- Dib, T.; Martinez-Pinto, J.; Reyes-Parada, M.; Torres, G.E.; Sotomayor-Zarate, R. Neonatal programming with testosterone propionate reduces dopamine transporter expression in nucleus accumbens and methylphenidate-induced locomotor activity in adult female rats. Behav. Brain Res. 2018, 346, 80–85. [Google Scholar] [CrossRef]
- Bonansco, C.; Martinez-Pinto, J.; Silva, R.A.; Velasquez, V.B.; Martorell, A.; Selva, M.V.; Espinosa, P.; Moya, P.R.; Cruz, G.; Andres, M.E.; et al. Neonatal exposure to oestradiol increases dopaminergic transmission in nucleus accumbens and morphine-induced conditioned place preference in adult female rats. J. Neuroendocrinol. 2018, 30, e12574. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: New York, NY, USA, 2009. [Google Scholar]
- Yorgason, J.T.; España, R.A.; Jones, S.R. Demon voltammetry and analysis software: Analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures. J. Neurosci. Methods 2011, 202, 158–164. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Thatcher, S.E.; Cassis, L.A. Measuring blood pressure using a noninvasive tail cuff method in mice. Methods Mol. Biol. 2017, 1614, 69–73. [Google Scholar] [PubMed]
- Volz, T.J.; Hanson, G.R.; Fleckenstein, A.E. The role of the plasmalemmal dopamine and vesicular monoamine transporters in methamphetamine-induced dopaminergic deficits. J. Neurochem. 2007, 101, 883–888. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Free Energy of Binding (Kcal/mol) | ||
|---|---|---|---|
| S1 | S2 | ||
| Cluster 1 | Cluster 2 | ||
| (±)-MTA | −6,4 | −5,5 | −5,3 |
| (±)-ETA | −6,4 | −5,5 | −5,4 |
| (±)-MT-But | −6,6 | −5,3 | −5,3 |
Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guajardo, F.G.; Velásquez, V.B.; Raby, D.; Núñez-Vivanco, G.; Iturriaga-Vásquez, P.; España, R.A.; Reyes-Parada, M.; Sotomayor-Zárate, R. Pharmacological Characterization of 4-Methylthioamphetamine Derivatives. Molecules 2020, 25, 5310. https://doi.org/10.3390/molecules25225310
Guajardo FG, Velásquez VB, Raby D, Núñez-Vivanco G, Iturriaga-Vásquez P, España RA, Reyes-Parada M, Sotomayor-Zárate R. Pharmacological Characterization of 4-Methylthioamphetamine Derivatives. Molecules. 2020; 25(22):5310. https://doi.org/10.3390/molecules25225310
Chicago/Turabian StyleGuajardo, Fabrizzio G., Victoria B. Velásquez, Daniela Raby, Gabriel Núñez-Vivanco, Patricio Iturriaga-Vásquez, Rodrigo A. España, Miguel Reyes-Parada, and Ramón Sotomayor-Zárate. 2020. "Pharmacological Characterization of 4-Methylthioamphetamine Derivatives" Molecules 25, no. 22: 5310. https://doi.org/10.3390/molecules25225310
APA StyleGuajardo, F. G., Velásquez, V. B., Raby, D., Núñez-Vivanco, G., Iturriaga-Vásquez, P., España, R. A., Reyes-Parada, M., & Sotomayor-Zárate, R. (2020). Pharmacological Characterization of 4-Methylthioamphetamine Derivatives. Molecules, 25(22), 5310. https://doi.org/10.3390/molecules25225310