
Application of New Efficient Hoveyda–Grubbs Catalysts Comprising an N→Ru Coordinate Bond in a Six-Membered Ring for the Synthesis of Natural Product-Like Cyclopenta[b]furo[2,3-c]pyrroles

 , ,
, ,  ,
, 
Abstract
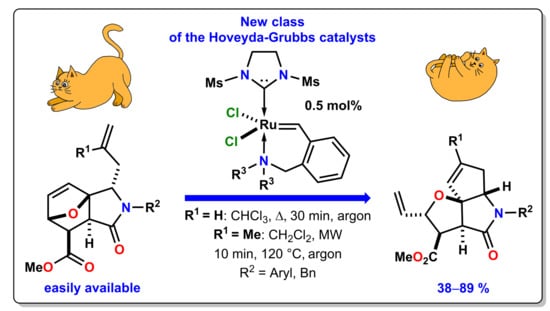
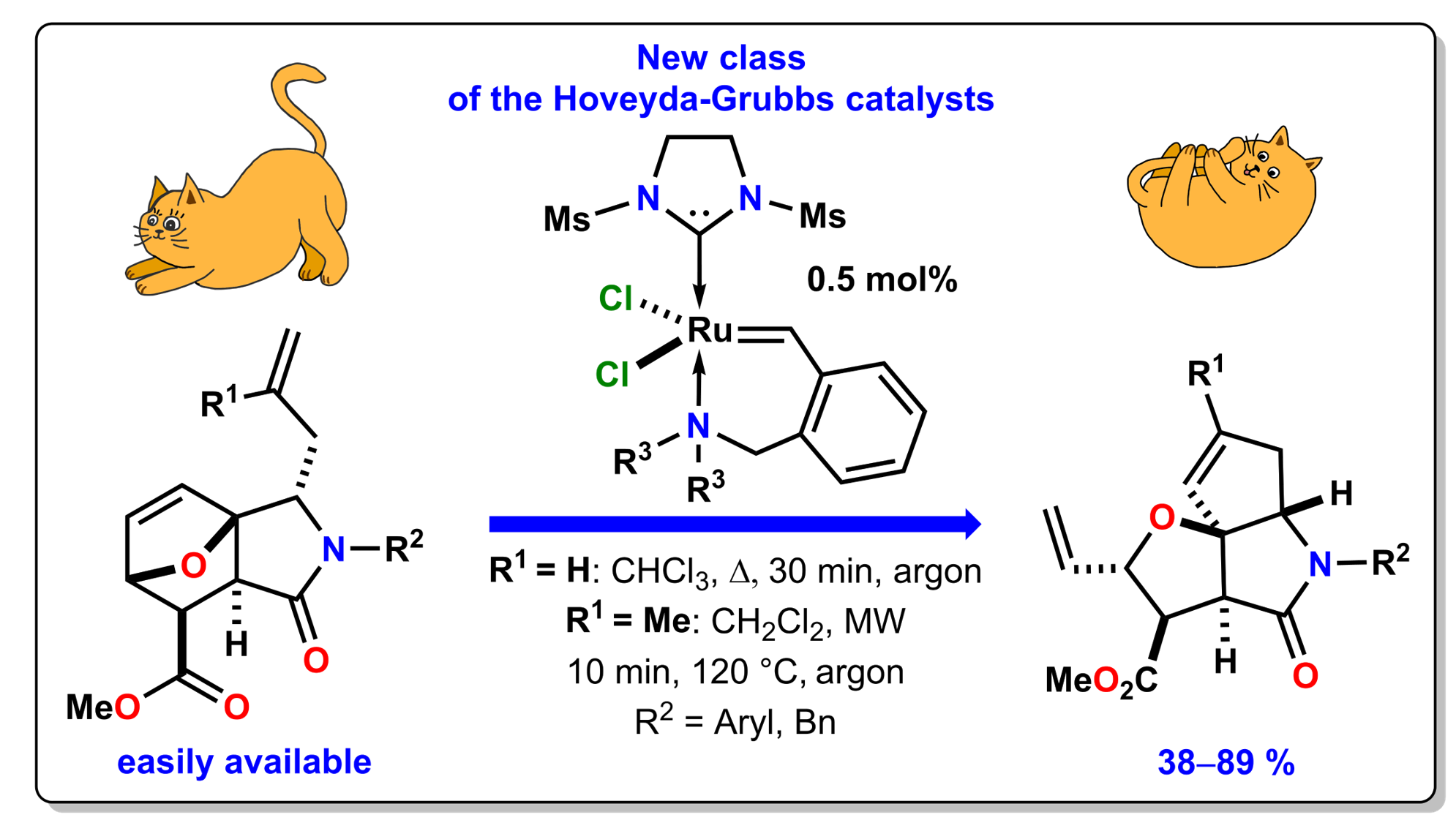
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Remarks
3.2. Experimental Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ogba, O.M.; Warner, N.C.; O’Leary, D.J.; Grubbs, R.H. Recent advances in ruthenium-based olefin metathesis. Chem. Soc. Rev. 2018, 47, 4510–4544. [Google Scholar] [CrossRef] [Green Version]
- Kotha, S.; Dipak, M.K. Strategies and tactics in olefin metathesis. Tetrahedron 2012, 68, 397–421. [Google Scholar] [CrossRef]
- Hughes, D.; Wheeler, P.; Ene, D. Olefin Metathesis in Drug Discovery and Development- Examples from Recent Patent Literature. Org. Process Res. Dev. 2017, 21, 1938–1962. [Google Scholar] [CrossRef]
- Higman, C.S.; Lummiss, J.A.; Fogg, D. Olefin Metathesis at the Dawn of Implementation in Pharmaceutical and Specialty-Chemicals Manufacturing. Angew. Chem. Int. Ed. 2016, 55, 3552–3565. [Google Scholar] [CrossRef]
- Yu, M.; Lou, S.; Gonzalez-Bobes, F. Closing Metathesis in Pharmaceutical Development: Fundamentals, Applications, and Future Directions. Org. Process Res. Dev. 2018, 22, 918–946. [Google Scholar] [CrossRef]
- Müller, D.S.; Baslé, O.; Mauduit, M.A. Tutorial review of stereoretentive olefin metathesis based on ruthenium dithiolate catalysts. Beilstein J. Org. Chem. 2018, 14, 2999–3010. [Google Scholar] [CrossRef]
- Montgomery, T.P.; Johns, A.M.; Grubbs, R.H. Recent Advancements in Stereoselective Olefin Metathesis Using Ruthenium Catalysts. Catalysts 2017, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Nolan, S.P.; Clavier, H. Chemoselective olefin metathesis transformations mediated by ruthenium complexes. Chem. Soc. Rev. 2010, 39, 3305–3316. [Google Scholar] [CrossRef]
- Lam, J.K.; Schmidt, Y.; Vanderwal, C.D. Complex polycyclic scaffolds by metathesis rearrangement of Himbert arene/allene cycloadducts. Org. Lett. 2012, 14, 5566–5569. [Google Scholar] [CrossRef]
- Kotha, S.; Meshram, M.; Khedkar, P.; Banerjee, S.; Deodhar, D. Recent applications of ring-rearrangement metathesis in organic synthesis. Beilstein J. Org. Chem. 2015, 11, 1833–1864. [Google Scholar] [CrossRef] [Green Version]
- Kotha, S.; Pulletikurti, S.A. Metathetic Approach to [5/5/6] Aza-Tricyclic Core of Dendrobine, Kopsanone, and Lycopalhine A Type of Alkaloids. Synthesis 2019, 51, 3981–3988. [Google Scholar] [CrossRef]
- Kotha, S.; Meshram, M.; Dommaraju, Y. Design and Synthesis of Polycycles, Heterocycles, and Macrocycles via Strategic Utilization of Ring-Closing Metathesis. Chem. Rec. 2018, 18, 1613–1632. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.; Kardos, M.; Vass, C.; Fülöp, F. Application of metathesis reactions in the synthesis and transformations of functionalized beta-amino acid derivatives. Synthesis 2018, 50, 3571–3588. [Google Scholar] [CrossRef] [Green Version]
- Acharyya, R.K.; Rej, R.K.; Nanda, S. Exploration of Ring Rearrangement Metathesis Reaction: A General and Flexible Approach for the Rapid Construction [5,n]-Fused Bicyclic Systems en Route to Linear Triquinanes. J. Org. Chem. 2018, 83, 2087–2103. [Google Scholar] [CrossRef] [PubMed]
- Clavier, H.; Broggi, J.; Nolan, S.P. Ring-Rearrangement Metathesis (RRM) Mediated by Ruthenium-Indenylidene Complexes. Eur. J. Org. Chem. 2010, 2010, 937–943. [Google Scholar] [CrossRef]
- Datta, R.; Ghosh, S. Domino ring-opening–ring-closing enyne metathesis vs. enyne metathesis of norbornene derivatives with alkynyl side chains. Construction of condensed polycarbocycles. Beilstein J. Org. Chem. 2018, 14, 2708–2714. [Google Scholar] [CrossRef] [Green Version]
- Kotha, S.; Ravikumar, O. Design and synthesis of oxa-bowls via Diels–Alder reaction and ring-rearrangement metathesis as key steps. Tetrahedron Lett. 2014, 55, 5781–5784. [Google Scholar] [CrossRef]
- Kotha, S.; Ravikumar, O. Ring-Rearrangement-Metathesis Approach to Polycycles: Substrate-Controlled Stereochemical Outcome During Grignard Addition. Eur. J. Org. Chem. 2016, 3900–3906. [Google Scholar] [CrossRef]
- Funel, J.A.; Prunet, J. Domino metathesis reactions for the synthesis of fused tricyclic frameworks. Synlett 2005, 235–238. [Google Scholar] [CrossRef]
- Kotha, S.; Ravikumar, O. Diversity−Oriented Approach to Carbocycles and Heterocycles through Ring− Rearrangement Metathesis, Fischer Indole Cyclization, and Diels—Alder Reaction as Key Steps. Eur. J. Org. Chem. 2014, 2014, 5582–5590. [Google Scholar] [CrossRef]
- Kotha, S.; Pulletikurti, S. Synthesis of propellanes containing a bicyclo[2.2.2]octene unit via the Diels–Alder reaction and ring-closing metathesis as key steps. RCS Adv. 2018, 8, 14906–14915. [Google Scholar] [CrossRef]
- Kotha, S.; Aswar, V.R. Target specific tactics in olefin metathesis: Synthetic approach to cis-syn-cis-triquinanes and propellanes. Org. Lett. 2016, 18, 1808–1811. [Google Scholar] [CrossRef] [PubMed]
- Cheng-Sánchez, I.; Sarabia, F. Recent Advances in Total Synthesis via Metathesis Reactions. Synthesis 2018, 50, 3749–3786. [Google Scholar]
- Firth, J.D.; Craven, P.G.E.; Lilburn, M.; Pahl, A.; Marsden, S.P.; Nelson, A. A biosynthesis-inspired approach to over twenty diverse natural product-like scaffolds. Chem. Commun. 2016, 52, 9837–9840. [Google Scholar] [CrossRef] [Green Version]
- Mariscal, R.; Maireles-Torres, P.; Ojeda, M.; Sádaba, I.; Granados, M.L. Furfural: A renewable and versatile platform molecule for the synthesis of chemicals and fuels. Energy Environ. Sci. 2016, 9, 1144–1189. [Google Scholar] [CrossRef]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Polyanskii, K.B.; Alekseeva, K.A.; Raspertov, P.V.; Kumandin, P.A.; Nikitina, E.V.; Gurbanov, A.V.; Zubkov, F.I. Hoveyda–Grubbs catalysts with an N→Ru coordinate bond in a six-membered ring. Synthesis of stable, industrially scalable, highly efficient ruthenium metathesis catalysts and 2-vinylbenzylamine ligands as their precursors. Beilstein J. Org. Chem. 2019, 15, 769–779. [Google Scholar] [CrossRef]
- Polyanskii, K.B.; Alekseeva, K.A.; Kumandin, P.A.; Atioğlu, Z.; Akkurt, M.; Toze, F.A. Crystal structure of [1,3-bis(2,4,6-trimethylphenyl)imidazolidin-2-ylidene]dichlorido{2-[1-(dimethylamino)ethyl]benzylidene} ruthenium including an unknown solvate. Acta Cryst. E 2019, 75, 342–345. [Google Scholar] [CrossRef]
- Zubkov, F.I.; Boltukhina, E.V.; Turchin, K.F.; Borisov, R.S.; Varlamov, A.V. New synthetic approach to substituted isoindolo[2,1-a]quinoline carboxylic acids via intramolecular Diels–Alder reaction of 4-(N-furyl-2)-4-arylaminobutenes-1 with maleic anhydride. Tetrahedron 2005, 61, 4099–4113. [Google Scholar] [CrossRef]
- Boltukhina, E.V.; Zubkov, F.I.; Nikitina, E.V.; Varlamov, A.V. Novel approach to isoindolo[2,1-a]quinolines: Synthesis of 1-and 3-halo-substituted 11-oxo-5,6,6a,11-tetrahydroisoindolo [2,1-a]quinoline-10-carboxylic acids. Synthesis 2005, 2005, 1859–1875. [Google Scholar] [CrossRef]
- Zubkov, F.I.; Boltukhina, E.V.; Nikitina, E.V.; Varlamov, A.V. Study of regioselectivity of intramolecular cyclization of N-(m-R-phenyl)- and N-(α-naphthyl)-2-allyl(methallyl)-6-carboxy-4-oxo-3-aza-10-oxatricyclo[5.2.1.01,5]dec-8-enes. Russ. Chem. Bull. 2004, 53, 2816–2829. [Google Scholar] [CrossRef]
- Varlamov, A.V.; Boltukhina, E.V.; Zubkov, F.I.; Nikitina, E.V.; Turchin, K.F. Intramolecular [4+2] cycloaddition of furfurylsubstituted homoallylamines to allylhalides, acryloyl chloride and maleic anhydride. J. Heterocyclic Chem. 2006, 43, 1479–1495. [Google Scholar] [CrossRef]
- Zubkov, F.I.; Nikitina, E.V.; Varlamov, A.V. Thermal and catalytic intramolecular [4+2]-cycloaddition in 2-alkenylfurans. Russ. Chem. Rev. 2005, 74, 639–669. [Google Scholar] [CrossRef]
- Padwa, A.; Flick, A.C. Intramolecular Diels–Alder Cycloaddition of Furans (IMDAF) for Natural Product Synthesis. Adv. Heterocycl. Chem. 2013, 110, 1–41. [Google Scholar]
- Kiselev, V.D.; Kornilov, D.A.; Sedov, I.A.; Konovalov, A.I. Solvent Influence on the Diels-Alder Reaction Rates of 9-(Hydroxymethyl)anthracene and 9,10-Bis(hydroxymethyl)anthracene with Two Maleimides. Int. J. Chem. Kinet. 2017, 49, 61–68. [Google Scholar] [CrossRef]
- Widstrom, A.L.; Lear, B.J. Structural and solvent control over activation parameters for a pair of retro Diels-Alder reactions. Sci. Rep. 2019, 9, 18267. [Google Scholar] [CrossRef]
- Gandini, A.; Coelho, D.; Silvestre, A.J. Reversible click chemistry at the service of macromolecular materials. Part 1: Kinetics of the Diels–Alder reaction applied to furan–maleimide model compounds and linear polymerizations. Eur. Polym. J. 2008, 44, 4029–4036. [Google Scholar] [CrossRef]
- Kotha, S.; Banerjee, S. Recent developments in the retro-Diels-Alder reaction. RSC Adv. 2013, 3, 7642–7666. [Google Scholar] [CrossRef]
- Pedrosa, R.; Andrés, C.; Nieto, J. A short diastereoselective synthesis of enantiopure highly substituted tetrahydroepoxyisoindolines. J. Org. Chem. 2000, 65, 831–839. [Google Scholar] [CrossRef]
- Zubkov, F.I.; Boltukhina, E.V.; Turchin, K.F.; Varlamov, A.V. An efficient approach to isoindolo [2,1-b][2] benzazepines via intramolecular [4+2] cycloaddition of maleic anhydride to 4-α-furyl-4-N-benzylaminobut-1-enes. Tetrahedron 2004, 60, 8455–8463. [Google Scholar] [CrossRef]
- Pedrosa, R.; Sayalero, S.; Vicente, M.; Casado, B. Chiral Template Mediated Diastereoselective Intramolecular Diels−Alder Reaction Using Furan as a Diene. Toward the Synthesis of Enantiopure Trisubstituted Tetrahydroepoxyisoindolones. J. Org. Chem. 2005, 70, 7273–7278. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Okada, T.; Taguchi, T.; Hanzawa, Y.; Iitaka, Y. Intramolecular Diels-Alder reactions of furan derivatives: Steric and electronic effects of trifluoromethyl groups. J. Fluorine Chem. 1992, 57, 239–243. [Google Scholar] [CrossRef]
- Holub, N.; Blechert, S. Ring-Rearrangement Metathesis. Chem. Asian J. 2007, 2, 1064–1082. [Google Scholar] [CrossRef] [PubMed]
- Harrity, J.P.; Visser, M.S.; Gleason, J.D.; Hoveyda, A.H. Ru-Catalyzed Rearrangement of Styrenyl Ethers. Enantioselective Synthesis of Chromenes through Zr-and Ru-Catalyzed Processes. J. Am. Chem. Soc. 1997, 119, 1488–1489. [Google Scholar] [CrossRef]
- Kouznetsov, V.; Öcal, N.; Turgut, Z.; Zubkov, F.; Kaban, S.; Varlamov, A. Allylation and Heterocycloaddition reactions of Aldimines: Furan- and Quinolinecarboxaldhydes. Monatsh. Chem. 1998, 129, 671–677. [Google Scholar] [CrossRef]
- Urbina, J.M.; Cortés, J.C.; Palma, A.; López, S.N.; Zacchino, S.A.; Enriz, R.D.; Ribas, C.; Kouznetsov, V.V. Inhibitors of the fungal cell wall. Synthesis of 4-aryl-4-N-arylamine-1-butenes and related compounds with inhibitory activities on β(1–3) glucan and chitin synthases. Bioorg. Med. Chem. 2000, 8, 691–698. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | Substituents | Conditions, Ratios of Trans-5A/cis-5B Isomers a, and Total Yields (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | CH2Cl2 −16 °C, 3 d | CH2Cl2 r.t, 3 d | MeCN r.t, 3 d | MeCN ∆, 3 h | PhH ∆, 3 h | PhMe r.t, 3 d | PhMe ∆, 3 h | |
| 5a | H | Ph | 76/24 (43) | 7/93 (58) | 47/53 (45) | 44/56 (15) | 59/41 (82) | 48/52 (85) | 59/41 (90) |
| 5b | H | 3-MeC6H4 | 63/37 (49) | 6/94 (65) | 59/44 (41) | 52/48 (25) | 58/42 (85) | 55/45 (90) | 57/43 (92) |
| 5g | H | 4-IC6H4 | 75/25 (52) | 0/100 (67) | 24/75 (38) | 28/72 (23) | 52/48 (86) | 78/22 (90) | 71/29 (91) |
| 5i | Me | Ph | 68/32 (63) | 23/77 (75) | 53/47 (62) | 50/50 (47) | 59/41 (95) | 67/33 (97) | 67/33 (98) |
| 5m | Me | Bn | 60/40 (64) | 4/96 (77) | 55/45 (65) | 58/42 (54) | 59/41 (96) | 59/41 (98) | 56/44 (99) |
| Average values | 68/32 (54) | 8/92 (68) | 48/52 (50) | 46/54 (33) | 57/43 (89) | 61/39 (92) | 62/38 (94) | ||
| Entry | R1 | R2 | Trans-5A/Cis-5B Ratio | Yield of Acid 5, % | Trans-6A/Cis-6B Ratio | Yield of Ester 6, % | Yield of Tricycle 7, % |
|---|---|---|---|---|---|---|---|
| a | H | Ph | 76/24 | 90 b | 79/21 | 85 | 68 c |
| 95/5 e | 53 e | 83 c | |||||
| 2/98 e | 12 e | 0 c | |||||
| b | H | 3-MeC6H4 | 57/43 | 95 a | 69/31 | 63 | 57 c |
| c | H | Bn | 90/10 | 57 b | 85/15 | 85 | 65 c |
| d | H | 3-ClC6H4 | 76/24 | 48 b | 70/30 | 80 | 57 c |
| e | H | 4-ClC6H4 | 80/20 | 91 a | 70/30 | 72 | 73 c |
| 98/2 e | 46 e | 89 c | |||||
| 8/92 e | 18 e | 0 c | |||||
| f | H | 4-BrC6H4 | 80/20 | 92 a | 72/28 | 70 | 63 c |
| 99/1 e | 46 e | 87 c | |||||
| 5/95 e | 12 e | 0 c | |||||
| g | H | 4-IC6H4 | 71/29 | 52 b | 59/41 | 63 | 48 c |
| h | H | 3-Cl,4-FC6H3 | 57/43 | 90 a | 66/34 | 86 | 54 c |
| i | Me | Ph | 68/32 | 63 b | 55/45 | 53 | 50 d |
| j | Me | 3-MeC6H4 | 60/40 | 67 b | 45/55 | 73 | 38 d |
| k | Me | 4-MeC6H4 | 51/49 | 93 a | 50/50 | 98 | 44 d |
| l | Me | 4-iPrC6H4 | 71/29 | 88 a | 87/13 | 82 | 62 d |
| m | Me | Bn | 56/44 | 64 a | 42/58 | 57 | 39 d |
| n | Me | 2-ClC6H4 | 71/29 | 63 b | 75/25 | 95 | 59 d |
| o | Me | 3-ClC6H4 | 68/32 | 69 b | 83/17 | 78 | 61 d |
| p | Me | 4-BrC6H4 | 76/24 | 64 b | 57/43 | 56 | 52 d |
| 65/35 e | 45 e | 61 d |
| Distances (Å) | Trans-6eA | Cis-6eB |
|---|---|---|
| H(3)…H(7a) | 3.781(1) | 2.915(1) |
| H(3)…H(4) | 3.493(1) | 2.871(1) |
| C(10) a…H(4) | 4.521(2) | 3.772(5) |
| Entry | Starting Ester | Catalyst (mol %) | Conditions | Target Tricycle | Yield, % a |
|---|---|---|---|---|---|
| 1 | 6eA | Cat. 1 (0.5) | CHCl3, Δ, 3 h, Ar | 7e | 85 |
| 2 | 6eA | Cat. 1 (0.5) | CHCl3, Δ, 1.5 h, Ar | 7e | 80 |
| 3 | 6eA | Cat. 1 (0.5) | CHCl3, Δ, 1.0 h, Ar | 7e | 82 |
| 4 | 6eA | Cat. 1 (0.5) | CHCl3, Δ, 0.5 h, Ar | 7e | 80 b |
| 5 | 6eA | HG-II (0.5) | CHCl3, Δ, 0.5 h, Ar | 7e | 74 |
| 6 | 6eA | Cat. 1 (0.5) | CHCl3, Δ, 0.25 h, air | 7e | 35 |
| 7 | 6eA | Cat. 1 (0.5) | CHCl3, Δ, 0.25 h, Ar | 7e | 83 b |
| 8 | 6eA | Cat. 1 (0.5) | CH2Cl2, Δ, 3 h, Ar | 7e | 58 |
| 9 | 6eA | Cat. 1 (0.5) | CH2Cl2, Δ, 1.5 h, Ar | 7e | 52 |
| 10 | 6eA | Cat. 1 (0.5) | CH2Cl2, Δ, 0.5 h, Ar | 7e | 37 |
| 11 | 6eA | Cat. 1 (1.0) | CHCl3, Δ, 0.25 h, Ar | 7e | 85 |
| 12 | 6eA | Cat. 1 (1.0) | CHCl3, Δ, 0.5 h, Ar | 7e | 85 |
| 13 | 6eA | Cat. 1 (0.1) | CHCl3, Δ, 0.25 h, Ar | 7e | 32 |
| 14 | 6eA | Cat. 1 (0.1) | CHCl3, Δ, 1.0 h, Ar | 7e | 51 |
| 15 | 6eA | Cat. 1 (0.1) | CHCl3, Δ, 1.5 h, Ar | 7e | 54 |
| 16 | 6eA | Cat. 1 (0.1) | CHCl3, Δ, 3 h, Ar | 7e | 55 |
| 17 | 6pA | Cat. 2 (0.5) | CHCl3, Δ, 30 min, Ar, MW, 120 °C | 7p | 55 |
| 18 | 6pA | Cat. 2 (0.5) | CHCl3, Δ, 20 min, Ar, MW, 120 °C | 7p | 52 |
| 19 | 6pA | Cat. 2 (0.5) | CHCl3, Δ, 10 min, Ar, MW, 120 °C | 7p | 45 |
| 20 | 6pA | Cat. 2 (0.5) | CH2Cl2, Δ, 30 min, Ar, MW, 120 °C | 7p | 85 |
| 21 | 6pA | Cat. 2 (0.5) | CH2Cl2, Δ, 20 min, Ar, MW, 120 °C | 7p | 84 |
| 22 | 6pA | Cat. 2 (0.5) | CH2Cl2, Δ, 15 min, Ar, MW, 120 °C | 7p | 80 |
| 23 | 6pA | Cat. 2 (0.5) | CH2Cl2, Δ, 10 min, Ar, MW, 100 °C | 7p | 0 |
| 24 | 6pA | Cat. 2 (0.5) | CH2Cl2, Δ, 10 min, Ar, MW, 120 °C | 7p | 82 b |
| 25 | 6pA | HG-II (0.5) | CH2Cl2, Δ, 10 min, Ar, MW, 120 °C | 7p | 42 |
| 26 | 6pA | Cat. 2 (1.0) | CH2Cl2, Δ, 10 min, Ar, MW, 120 °C | 7p | 83 |
| 25 | 6pA | Cat. 2 (1.0) | CH2Cl2, Δ, 15 min, Ar, MW, 120 °C | 7p | 85 |
| 26 | 6pA | Cat. 2 (0.1) | CH2Cl2, Δ, 10 min, Ar, MW, 120 °C | 7p | 43 |
| 27 | 6pA | Cat. 2 (0.1) | CH2Cl2, Δ, 15 min, Ar, MW, 120 °C | 7p | 47 |
Sample Availability: Samples of the compounds 5–7 are available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antonova, A.S.; Vinokurova, M.A.; Kumandin, P.A.; Merkulova, N.L.; Sinelshchikova, A.A.; Grigoriev, M.S.; Novikov, R.A.; Kouznetsov, V.V.; Polyanskii, K.B.; Zubkov, F.I. Application of New Efficient Hoveyda–Grubbs Catalysts Comprising an N→Ru Coordinate Bond in a Six-Membered Ring for the Synthesis of Natural Product-Like Cyclopenta[b]furo[2,3-c]pyrroles. Molecules 2020, 25, 5379. https://doi.org/10.3390/molecules25225379
Antonova AS, Vinokurova MA, Kumandin PA, Merkulova NL, Sinelshchikova AA, Grigoriev MS, Novikov RA, Kouznetsov VV, Polyanskii KB, Zubkov FI. Application of New Efficient Hoveyda–Grubbs Catalysts Comprising an N→Ru Coordinate Bond in a Six-Membered Ring for the Synthesis of Natural Product-Like Cyclopenta[b]furo[2,3-c]pyrroles. Molecules. 2020; 25(22):5379. https://doi.org/10.3390/molecules25225379
Chicago/Turabian StyleAntonova, Alexandra S., Marina A. Vinokurova, Pavel A. Kumandin, Natalia L. Merkulova, Anna A. Sinelshchikova, Mikhail S. Grigoriev, Roman A. Novikov, Vladimir V. Kouznetsov, Kirill B. Polyanskii, and Fedor I. Zubkov. 2020. "Application of New Efficient Hoveyda–Grubbs Catalysts Comprising an N→Ru Coordinate Bond in a Six-Membered Ring for the Synthesis of Natural Product-Like Cyclopenta[b]furo[2,3-c]pyrroles" Molecules 25, no. 22: 5379. https://doi.org/10.3390/molecules25225379
APA StyleAntonova, A. S., Vinokurova, M. A., Kumandin, P. A., Merkulova, N. L., Sinelshchikova, A. A., Grigoriev, M. S., Novikov, R. A., Kouznetsov, V. V., Polyanskii, K. B., & Zubkov, F. I. (2020). Application of New Efficient Hoveyda–Grubbs Catalysts Comprising an N→Ru Coordinate Bond in a Six-Membered Ring for the Synthesis of Natural Product-Like Cyclopenta[b]furo[2,3-c]pyrroles. Molecules, 25(22), 5379. https://doi.org/10.3390/molecules25225379