3. Materials and Methods
3.1. Chemistry
All reagents were obtained from commercial suppliers and were used without any further purification unless otherwise stated. Flash column chromatography was performed with silica gel (200-300 mesh) purchased from Qingdao Haiyang Chemical Co. Ltd. Thin layer chromatography was performed using silica gel 60 F254 precoated plates (purchased from Qingdao Haiyang Inc., Qingdao, China). Visualization was achieved using Ultraviolet (UV) light (254 nm and 365 nm, Shanghai Yarong Biochemical Instrument Factory, Shanghai, China). Melting points were determined with a Mel-TEMP II melting point apparatus (Beijing Keyi Company, Beijing, China) and was uncorrected. 1H NMR and 13C NMR spectra were recorded with Bruker AV-600, AV-500 or AV-400 MHz instruments (Bruker, Ettlingen, Germany) using DMSO-d6, CD3OD, or CDCl3 as solvent. Chemical shifts were reported as δ values (ppm) from internal reference tetramethylsilane (TMS). All coupling constants were reported in hertz (Hz), All chemical shifts are reported in parts per million (ppm), relative to the internal standard. In addition, proton multiplicities were labeled as br (broad), s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), and m (multiplet). HR-MS were performed on a Waters Vion IMS Q-tof (Waters, MA, USA).
3.2. General Procedure for the Synthesis of Compounds 3a–d
4-Piperidinecarboxamide (1) (3.00 g, 23.4 mmol) and substituted Benzylchloride derivatives (2a–d) (28.1 mmol) were dissolved in 20 mL acetone. Then, anhydrous K2CO3 (6.47 g, 46.8 mmol) and catalytic amount KI were added. The reaction mixture was refluxed for 4 h. After completion of the reaction, acetone was concentrated, and the residue was dissolved in water (60 mL) and extracted with ethyl acetate (60 × 3 mL). The combined organic layers were dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. After concentration, the crude product was purified by silica gel column chromatograph (DCM: methanol = 60:1–5:1) to give target compounds 3a–d.
3.3. General Procedure for the Synthesis of Compounds 4a–d
Compounds 3a–d (3 g) were dissolved in anhydrous THF (17 mL) and then LiAlH4 (5 equiv.) was added to the above cooled solution at 0–5 °C in small portions under stirring. The reaction mixture was further stirred at room temperature for 30 min and finally refluxed for 4 h. After cooling, water and 10% NaOH solution was added at 0–5 °C. Then, the obtained white precipitate was filtered off and washed with THF. The filtrate was extracted with ethyl acetate. The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The obtained compounds 4a–d were used in further synthesis without purification.
3.4. General Procedure for the Synthesis of Compounds 6a–h
CDI (1 equiv.) was added to a solution of the acids (5a–d) ((300 mg, 1 equiv.) in dry THF under nitrogen atmosphere. After 30 min, the solution of substituted 4-Amine-1-benzylpiperidines (4a–d) (1.2 equiv.) in THF were added, and the reaction mixture was stirred at room temperature for 24 h. After the reaction was completed, the solvent was removed under reduced pressure, and then the reaction mixture was quenched with saturated NaCl solution (25 mL). The aqueous phase was extracted with DCM (25 × 3 mL). The DCM layer was combined and washed with brine solution (25 × 3 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. After concentration, the crude product was purified by silica gel column chromatograph (DCM: methanol = 60:1–5:1) to give target compounds 6a–h.
N-((1-Benzylpiperidin-4-yl)methyl)-1H-indole-5-carboxamide (6a). 1H-Indole-5-carboxylic acid (300 mg, 1.86 mmol), CDI (302 mg, 1.86 mmol), (1-Benzylpiperidin-4-yl)methanamine (457 mg, 2.24 mmol), THF (15 mL) White solid, m.p.: 89–90 °C, yield: 80%, 1H NMR (500 MHz, DMSO-d6) δ 11.50 (s, 1H), 8.47 (s, 1H), 8.17 (s, 1H), 7.66 (d, J = 8.4 Hz, 1H), 7.45 (d, J = 8.6 Hz, 1H), 7.41 (d, J = 7.1 Hz, 2H), 7.36–7.29 (m, 3H), 6.51 (s, 1H), 3.78 (s, 2H), 3.19 (d, J = 5.5 Hz, 2H), 2.99 (d, J = 10.8 Hz, 2H), 2.31 (d, J = 11.0 Hz, 2H), 1.72 (d, J = 12.0 Hz, 3H), 1.38 (d, J = 11.4 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 168.15, 137.84, 135.07, 130.40, 128.81, 128.39, 127.44, 127.05, 125.94, 120.97, 120.37, 111.37, 102.49, 61.15, 52.37, 44.70, 35.20, 28.63. HRMS (ESI): calcd. For C22H25N3O [M + H]+ 348.2070, found 348.2070.
N-((1-(2-Chlorobenzyl)piperidin-4-yl)methyl)-1H-Indole-5-carboxamide (6b). 1H-Indole-5-carboxylic acid (300 mg, 1.86 mmol), CDI (302 mg, 1.86 mmol), (1-(2-Chlorobenzyl) piperidin-4-yl)methanamine (532 mg, 2.24 mmol), THF (15 mL) White solid, m.p.: 88–89 °C, yield: 66%, 1H NMR (400 MHz, CD3OD) δ 8.08 (d, J = 1.1 Hz, 1H), 7.58 (dd, J = 8.6, 1.8 Hz, 1H), 7.49 (dd, J = 7.1, 2.2 Hz, 1H), 7.41–7.35 (m, 2H), 7.30–7.24 (m, 3H), 6.51 (d, J = 3.2 Hz, 1H), 3.78 (s, 2H), 3.05 (d, J = 11.7 Hz, 2H), 2.31 (t, J = 11.9 Hz, 2H), 1.77 (t, J = 14.0 Hz, 3H), 1.45–1.33 (m, 2H). 13C NMR (101 MHz, CD3OD) δ 170.60, 138.15, 134.64, 131.71, 129.37, 129.10, 127.70, 126.78, 125.91, 125.17, 120.18, 119.95, 110.67, 102.17, 58.57, 53.06, 44.79, 35.56, 28.93. HRMS (ESI): calcd. For C22H24ClN3O [M + H]+ 382.1681, found 382.1703.
N-((1-Benzylpiperidin-4-yl) methyl)-1H-indazole-5-carboxamide (6c). 1H-Indazole-5-carboxylic acid (300 mg, 1.86 mmol), CDI (302 mg, 1.86 mmol), (1-Benzylpiperidin-4-yl)methanamine (457 mg, 2.24 mmol), THF (15 mL) White solid, m.p.: 111–112 °C, yield: 56%, 1H NMR (500 MHz, DMSO-d6) δ 13.30 (s, 1H), 8.48 (s, 1H), 8.35 (s, 1H), 8.20 (s, 1H), 7.87 (d, J = 8.8 Hz, 1H), 7.57 (d, J = 8.7 Hz, 1H), 7.33–7.27 (m, 3H), 7.24 (d, J = 6.8 Hz, 1H), 3.45 (s, 2H), 3.18 (d, J = 6.0 Hz, 2H), 2.81 (d, J = 11.1 Hz, 2H), 1.92 (t, J = 11.1 Hz, 2H), 1.67 (d, J = 12.4 Hz, 2H), 1.59 (s, 1H), 1.22 (d, J = 9.4 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 167.07, 141.33, 138.83, 135.11, 129.24, 128.56, 127.67, 127.62, 127.30, 125.79, 122.79, 120.90, 110.16, 62.78, 53.38, 45.31, 36.17, 30.23. HRMS (ESI): calcd. For C21H24N4O [M + H]+ 349.2023, found 349.2019.
N-((1-Benzylpiperidin-4-yl)methyl)-1H-benzo[d]imidazole-5-carboxamide (6d). 1H-Benzo[d]imidazole-5-carboxylic acid (300 mg, 1.85 mmol), CDI (300 mg, 1.85 mmol), (1-Benzylpiperidin-4-yl)methanamine (453 mg, 2.22 mmol), THF (15 mL) Yellow oil, yield: 72%, 1H NMR (500 MHz, DMSO-d6) δ 8.53 (s, 1H), 8.27 (d, J = 68.8 Hz, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.70–7.61 (m, 1H), 7.28 (d, J = 6.9 Hz, 3H), 7.21 (d, J = 5.6 Hz, 1H), 7.04 (s, 1H), 3.39 (s, 2H), 3.19 (s, 2H), 2.76 (s, 2H), 1.85 (d, J = 11.8 Hz, 2H), 1.65 (d, J = 11.8 Hz, 3H), 1.20 (d, J = 10.9 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 167.40, 144.24, 139.08, 135.64, 129.16, 128.51, 127.18, 121.88, 115.66, 114.85, 62.93, 53.43, 53.35, 45.39, 36.25, 30.32. HRMS (ESI): calcd. For C21H24N4O [M + H]+ 349.2023, found 349.2023.
N-((1-(4-(Trifluoromethyl)benzyl)piperidin-4-yl)methyl)-1H-benzo[d]imidazole-5-carboxamide (6e). 1H-Benzo[d]imidazole-5-carboxylic acid (300 mg, 1.85 mmol), CDI (300 mg, 1.85 mmol), (1-(4-(Trifluoromethyl)benzyl)piperidin-4-yl)methanamine (604 mg, 2.22 mmol), THF (15 mL) White solid, m.p.: 92–93 °C, yield: 77%, 1H NMR (400 MHz, CD3OD) δ 8.29–8.27 (m, 1H), 8.12 (d, J = 1.0 Hz, 1H), 7.87–7.77 (m, 2H), 7.62 (d, J = 7.8 Hz, 1H), 7.56 (t, J = 8.9 Hz, 2H), 7.39 (p, J = 5.5 Hz, 2H), 3.66 (d, J = 6.7 Hz, 2H), 3.30 (s, 1H), 3.27 (s, 1H), 2.90 (d, J = 11.4 Hz, 2H), 2.15–2.06 (m, 2H), 1.79–1.63 (m, 3H), 1.44–1.24 (m, 3H). 13C NMR (126 MHz, CD3OD) δ 169.23, 141.32, 134.81, 131.92, 130.75, 130.34, 128.00, 127.37, 127.30, 125.41, 122.55, 120.67, 114.13, 113.58, 109.69, 58.94, 57.96, 53.34, 53.01, 44.93, 44.83, 35.64, 35.52, 29.26, 29.06. HRMS (ESI): calcd. For C22H23F3N4O [M + H]+ 417.1897, found 417.1895.
N-((1-(4-Methoxybenzyl)piperidin-4-yl)methyl)-1H-benzo[d][1,2,3]triazole-5-carboxamide (6f). 1H-Benzo[d][1,2,3]triazole-5-carboxylic acid (300 mg, 1.84 mmol), CDI (298 mg, 1.84 mmol), (1-(4-Methoxybenzyl)piperidin-4-yl)methanamine (517 mg, 2.21 mmol), THF (15 mL) Yellow oil, yield: 63%, 1H NMR (500 MHz, DMSO-d6) δ 8.53 (s, 1H), 8.32 (s, 1H), 8.16 (s, 1H), 7.75 (d, J = 8.4 Hz, 1H), 7.61 (d, J = 8.3 Hz, 1H), 7.32 (d, J = 8.3 Hz, 2H), 6.91 (d, J = 8.3 Hz, 2H), 3.74 (s, 3H), 3.69 (s, 2H), 3.18 (d, J = 5.9 Hz, 2H), 2.97 (d, J = 11.1 Hz, 2H), 2.26 (s, 2H), 1.73 (d, J = 13.0 Hz, 2H), 1.68 (s, 1H), 1.34 (d, J = 11.2 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 167.31, 159.33, 144.22, 131.57, 128.92, 121.94, 114.16, 60.78, 55.52, 52.34, 44.84, 35.34, 28.86. HRMS (ESI): calcd. For C21H25N5O2 [M + H]+ 380.2081, found 380.2077.
N-((1-(2-Chlorobenzyl)piperidin-4-yl)methyl)-1H-benzo[d][1,2,3]triazole-5-carboxamide (6g). 1H-Benzo[d][1,2,3]triazole-5-carboxylic acid (300 mg, 1.84 mmol), CDI (298 mg, 1.84 mmol), (1-(2-Chlorobenzyl)piperidin-4-yl)methanamine (526 mg, 2.21 mmol), THF (15 mL) White solid, m.p.: 77–79 °C, yield: 57%, 1H NMR (500 MHz, DMSO-d6) δ 8.63 (s, 1H), 8.44 (s, 1H), 7.87 (d, J = 2.7 Hz, 2H), 7.47 (d, J = 7.1 Hz, 1H), 7.39 (d, J = 7.8 Hz, 1H), 7.30 (d, J = 6.9 Hz, 1H), 7.27–7.25 (m, 1H), 7.03 (s, 1H), 3.52 (s, 2H), 3.21 (t, J = 6.1 Hz, 2H), 2.82 (d, J = 11.3 Hz, 2H), 2.00 (t, J = 10.4 Hz, 2H), 1.68 (d, J = 12.0 Hz, 2H), 1.61 (s, 1H), 1.23 (d, J = 10.5 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 166.50, 140.30, 139.38, 138.70, 131.96, 129.27, 128.56, 127.32, 125.46, 115.61, 114.32, 62.73, 53.33, 45.41, 36.08, 30.16. HRMS (ESI): calcd. For C20H22ClN5O [M + H]+ 384.1586, found 384.1584.
N-((1-(4-(Trifluoromethyl)benzyl)piperidin-4-yl)methyl)-1H-benzo[d][1,2,3]triazole-5-carboxamide (6h). 1H-Benzo[d][1,2,3]triazole-5-carboxylic acid (300 mg, 1.84 mmol), CDI (298 mg, 1.84 mmol), (1-(4-(Trifluoromethyl)benzyl)piperidin-4-yl)methanamine (601 mg, 2.21 mmol), THF (15 mL) White solid, m.p.: 90–91 °C, yield: 73%, 1H NMR (400 MHz, CD3OD) δ 8.38 (td, J = 1.5, 1.0 Hz, 1H), 7.92–7.90 (m, 1H), 7.88 (t, J = 1.0 Hz, 1H), 7.81 (d, J = 7.8 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.61 (dt, J = 7.7, 4.1 Hz, 1H), 7.51–7.42 (m, 2H), 3.89 (d, J = 22.6 Hz, 2H), 3.33 (dd, J = 6.5, 3.7 Hz, 2H), 3.13–3.01 (m, 2H), 2.47–2.31 (m, 2H), 1.90–1.68 (m, 3H), 1.52–1.32 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 166.50, 140.30, 139.38, 138.70, 131.96, 129.27, 128.56, 127.32, 125.46, 115.61, 114.32, 62.73, 53.33, 45.41, 36.08, 30.16. HRMS (ESI): calcd. For C21H22F3N5O [M + H]+ 418.1849, found 418.1850.
3.5. General Procedure for the Synthesis of 8i–l
Intermediates (7) (140 mg, 1.2 equiv.), PyBOP (1.2 equiv.) and DIPEA (1.5 equiv.) were added to DMF and stirred at room temperature for 20 min. Then, intermediates (4a–d) (1.0 equiv.) was added and stirred at room temperature for 4 h. After completion of the reaction, the reaction mixture was quenched with saturated NaCl solution (25 mL). The aqueous phase was extracted with DCM (25 × 3 mL). The DCM layer was combined and washed with brine solution (25 × 3 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. After concentration, the crude product was purified by silica gel column chromatograph using a methanol in DCM gradient (DCM: methanol= 60:1–5:1) yielded compounds 8i–l.
N-((1-Benzylpiperidin-4-yl)methyl)-2-Oxoindoline-5-carboxamide (8i). 2-Oxoindoline-5-carboxylic acid (140 mg, 0.79 mmol), (1-Benzylpiperidin-4-yl)methanamine (135 mg, 0.66 mmol), PyBOP (412 mg, 0.79 mmol), DIPEA(128 mg, 0.99 mmol), DMF (6 mL). White solid, m.p.:155–156 °C, yield: 56%, 1H NMR (400 MHz, CD3OD) δ 7.73–7.68 (m, 2H), 7.53–7.42 (m, 5H), 6.91 (d, J = 8.1 Hz, 1H), 4.26 (s, 2H), 3.44 (d, J = 12.5 Hz, 2H), 3.31 (s, 1H), 2.98 (t, J = 12.9 Hz, 2H), 1.97 (d, J = 13.6 Hz, 3H), 1.54 (q, J = 13.1, 12.1 Hz, 2H). 13C NMR (101 MHz, CD3OD) δ 178.52, 168.90, 146.69, 130.99, 129.80, 129.40, 128.99, 127.92, 127.60, 125.95, 123.42, 123.40, 108.97, 60.21, 51.86, 43.82, 42.11, 34.01, 26.82. HRMS (ESI): calcd. For C22H25N3O2 [M + H]+ 364.2020, found 364.2032.
N-((1-(2-Chlorobenzyl)piperidin-4-yl)methyl)-2-Oxoindoline-5-carboxamide (8j). 2-Oxoindoline-5-carboxylic acid (140 mg, 0.79 mmol), (1-(2-Chlorobenzyl)piperidin-4-yl)methanamine (157 mg, 0.66 mmol), PyBOP (412 mg, 0.79 mmol), DIPEA(128 mg, 0.99 mmol), DMF (6 mL). White solid, m.p.: 151–152 °C, yield: 35%, 1H NMR (600 MHz, CD3OD) δ 7.74–7.69 (m, 2H), 7.50 (d, J = 7.2 Hz, 1H), 7.39 (d, J = 7.6 Hz, 1H), 7.28 (dt, J = 20.1, 7.1 Hz, 2H), 6.93 (d, J = 8.1 Hz, 1H), 3.71 (s, 2H), 3.27 (d, J = 6.8 Hz, 2H), 3.00 (s, 2H), 2.21 (s, 2H), 1.76 (d, J = 12.7 Hz, 2H), 1.69 (s, 1H), 1.37 (q, J = 11.5 Hz, 2H). 13C NMR (101 MHz, CD3OD) δ 168.83, 146.63, 135.02, 132.40, 130.20, 129.68, 128.07, 127.58, 127.18, 125.93, 123.41, 108.96, 57.94, 52.76, 44.36, 34.83, 28.01. HRMS (ESI): calcd. For C22H24ClN3O2 [M + H]+ 398.1630, found 398.1652.
N-((1-(4-Methoxybenzyl)piperidin-4-yl)methyl)-2-Oxoindoline-5-carboxamide (8k). 2-Oxoindoline-5-carboxylic acid (140 mg, 0.79 mmol), (1-(4-Methoxybenzyl)piperidin-4-yl)methanamine (154 mg, 0.66 mmol), PyBOP (412 mg, 0.79 mmol), DIPEA(128 mg, 0.99 mmol), DMF (6 mL). White solid, m.p.: 158–159 °C, yield: 43%, 1H NMR (400 MHz, CD3OD) δ 7.74–7.68 (m, 2H), 7.40 (d, J = 8.7 Hz, 2H), 6.98 (d, J = 8.7 Hz, 2H), 6.91 (d, J = 8.0 Hz, 1H), 4.19 (s, 2H), 3.79 (s, 3H), 3.43 (d, J = 12.0 Hz, 2H), 3.31 (s, 1H), 2.95 (t, J = 11.7 Hz, 2H), 1.97 (d, J = 13.9 Hz, 3H), 1.60–1.45 (m, 2H), 1.26 (s, 1H). 13C NMR (101 MHz, CD3OD) δ 178.54, 168.87, 161.10, 146.66, 132.51, 127.95, 127.62, 125.93, 123.46, 121.09, 114.21, 108.98, 54.55, 51.51, 43.78, 34.03, 27.22, 26.85. HRMS (ESI): calcd. For C23H27N3O3 [M + H]+ 394.2125, found 394.2131.
2-Oxo-N-((1-(4-(trifluoromethyl)benzyl)piperidin-4-yl)methyl)indoline-5-carboxamide (8l). 2-Oxoindoline-5-carboxylic acid (140 mg, 0.79 mmol), (1-(4-(Trifluoromethyl)benzyl)piperidin-4-yl)methanamine (179 mg, 0.65 mmol), PyBOP (412 mg, 0.79 mmol), DIPEA(128 mg, 0.99 mmol), DMF (6 mL). White solid, m.p.: 113–115 °C, yield: 40%, 1H NMR (400 MHz, CD3OD) δ 7.79 (d, J = 7.8 Hz, 1H), 7.73–7.69 (m, 2H), 7.62 (d, J = 8.2 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44–7.32 (m, 2H), 6.95–6.90 (m, 1H), 3.60 (d, J = 17.7 Hz, 2H), 3.24 (dd, J = 9.1, 6.8 Hz, 2H), 2.91–2.77 (m, 2H), 2.09–1.93 (m, 2H), 1.78–1.61 (m, 3H), 1.40–1.21 (m, 3H). 13C NMR (126 MHz, CD3OD)) δ 168.70, 146.44, 131.73, 130.35, 130.30, 128.23, 127.47, 127.30, 126.79, 125.81, 125.26, 123.34, 108.90, 59.79, 59.79, 58.24, 53.43, 53.14, 45.07, 36.04, 36.04, 36.00, 36.00, 29.82, 29.76. HRMS (ESI): calcd. For C23H24F3N3O2 [M + H]+ 432.1893, found 432.1886.
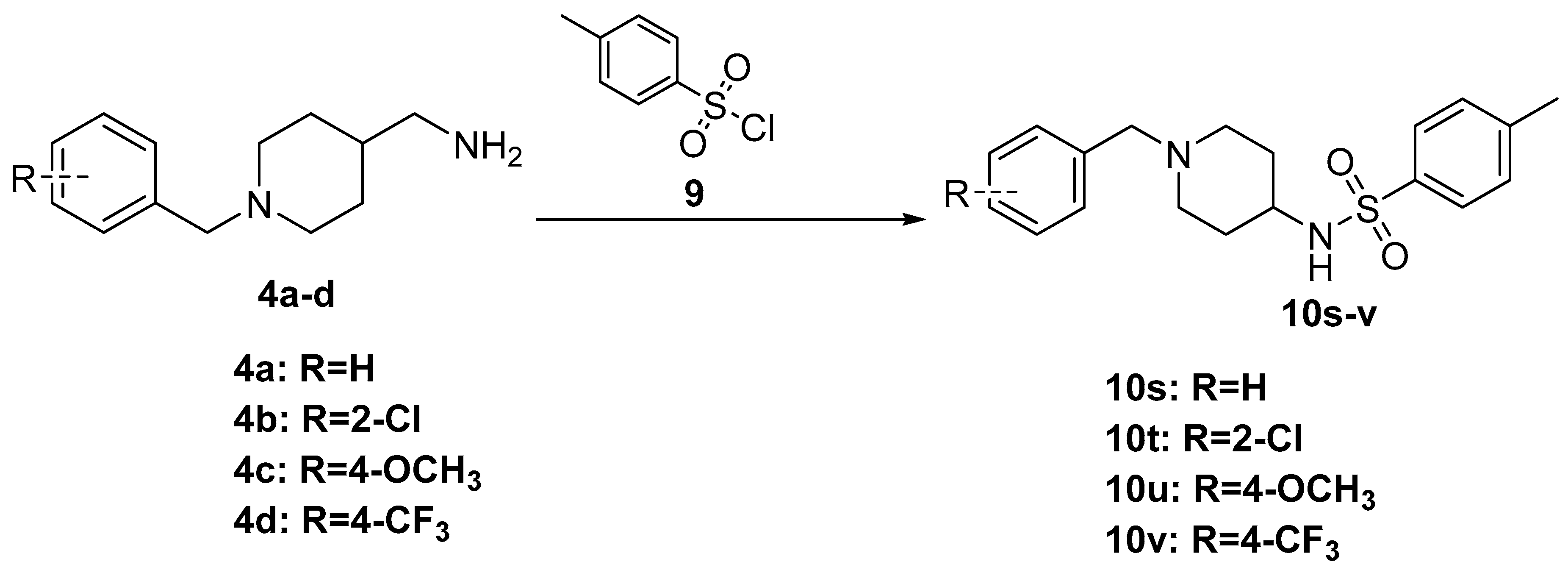
3.6. General Procedure for the Synthesis of 10s–v
To a solution of (1-Benzylpiperidin-4-yl) methanamine derivatives (4a–d) (150 mg, 1 equiv.) in DCM (5 mL) was added Et3N (1.8 equiv.). The mixture was cooled to 0 °C, TsCl (1.3 equiv.) in DCM (5 mL) was added dropwise, and the reaction mixture was stirred for an additional 1 h at 0 °C. After completion of the reaction, the reaction mixture was dissolved in 15 mL saturated sodium bicarbonate (NaHCO3) and extracted with DCM (25 × 3 mL). Organic phases were combined and washed with saturated NaCl solution (25 × 3 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. After concentration, the crude product was purified by silica gel column chromatograph using a methanol in dichloromethane gradient (dichloromethane: methanol = 60:1–5:1) yielded compounds 10s–v.
N-((1-Benzylpiperidin-4-yl)methyl)-4-methylbenzenesulfonamide (10s). (1-Benzylpiperidin-4-yl) methanamine (150 mg, 0.73 mmol), TsCl (181 mg, 0.95 mmol), Et3N (133 mg, 1.31 mmol), DCM (9 mL) White solid, m.p.:78–79 °C, yield: 80%, 1H NMR (400 MHz, CD3OD) δ 7.68 (d, J = 8.3 Hz, 2H), 7.36–7.24 (m, 7H), 3.60 (s, 2H), 2.93 (d, J = 12.0 Hz, 2H), 2.67 (d, J = 6.8 Hz, 2H), 2.39 (s, 3H), 2.09 (t, J = 12.6 Hz, 2H), 1.68 (d, J = 13.8 Hz, 2H), 1.44 (s, 1H), 1.28–1.13 (m, 2H). 13C NMR (101 MHz, CD3OD) δ 143.22, 137.71, 136.09, 129.69, 129.36, 128.05, 127.39, 126.67, 62.59, 52.72, 35.66, 28.79, 20.11. HRMS (ESI): calcd. For C20H26N2O2S [M + H]+ 359.1788, found 359.1802.
N-((1-(2-Chlorobenzyl)piperidin-4-yl)methyl)-4-methylbenzenesulfonamide (10t). (1-(4-Chlorobenzyl)piperidin-4-yl)methanamine (150 mg,0.63 mmol), TsCl (156 mg, 0.82 mmol), Et3N (114 mg, 1.13 mmol), DCM (9 mL) White solid, m.p.: 111–112 °C, yield: 73%, 1H NMR (400 MHz, CD3OD) δ 7.69 (d, J = 8.3 Hz, 2H), 7.44 (dd, J = 7.4, 2.0 Hz, 1H), 7.37–7.31 (m, 3H), 7.23 (td, J = 7.1, 1.9 Hz, 2H), 3.62 (s, 2H), 2.89 (d, J = 11.8 Hz, 2H), 2.67 (d, J = 6.8 Hz, 2H), 2.39 (s, 3H), 2.06 (t, J = 11.7 Hz, 2H), 1.65 (d, J = 13.1 Hz, 2H), 1.37 (s, 1H), 1.20 (dd, J = 12.3, 3.6 Hz, 1H), 1.15 (s, 1H). 13C NMR (101 MHz, CD3OD) δ 143.42, 137.49, 135.59, 133.53, 131.79, 130.10, 129.48, 127.74, 127.35, 126.68, 126.68, 20.13. HRMS (ESI): calcd. For C20H25ClN2O2S [M + H]+ 393.1398, found 393.1397.
N-((1-(4-Methoxybenzyl)piperidin-4-yl)methyl)-4-methylbenzenesulfonamide (10u). (1-(4-Methoxybenzyl)piperidin-4-yl)methanamine (150 mg, 0.64 mmol), TsCl (158 mg, 0.83 mmol), Et3N (117 mg, 1.15 mmol), DCM (9 mL) White solid, m.p.: 76–77 °C, yield: 87%, 1H NMR (600 MHz, CD3OD) δ 7.61 (d, J = 8.3 Hz, 2H), 7.25 (dd, J = 27.4, 8.3 Hz, 4H), 6.85 (d, J = 8.6 Hz, 2H), 3.80 (s, 2H), 3.70 (s, 3H), 3.10 (d, J = 10.7 Hz, 2H), 2.63 (d, J = 6.7 Hz, 2H), 2.44 (s, 2H), 2.32 (s, 3H), 2.05 (s, 1H), 1.73 (s, 2H), 1.52 (s, 1H), 1.27–1.19 (m, 2H). 13C NMR (101 MHz, CD3OD) δ 160.41, 143.33, 137.60, 131.82, 129.42, 126.66, 113.87, 60.67, 54.47, 51.88, 34.72, 27.47, 20.10. HRMS (ESI): calcd. For C21H28N2O3S [M + H]+ 389.1893, found 389.1894.
4-Methyl-N-((1-(4-(trifluoromethyl)benzyl)piperidin-4-yl)methyl)benzenesulfonamide (10v). (1-(4-(Trifluoromethyl)benzyl)piperidin-4-yl)methanamine (150 mg, 0.55 mmol), TsCl (136 mg, 0.72 mmol), Et3N (100 mg, 0.99 mmol), DCM (9 mL) White solid, m.p.: 87–88 °C, yield: 88%, 1H NMR (400 MHz, CD3OD) δ 7.76 (d, J = 7.8 Hz, 1H), 7.71–7.66 (m, 2H), 7.61 (d, J = 7.9 Hz, 1H), 7.57–7.51 (m, 1H), 7.40–7.31 (m, 4H), 3.58 (d, J = 14.0 Hz, 2H), 2.79 (t, J = 12.2 Hz, 2H), 2.67 (t, J = 6.9 Hz, 2H), 2.40 (s, 3H), 1.97 (t, J = 11.6 Hz, 2H), 1.69–1.59 (m, 2H), 1.40 (dtt, J = 14.5, 7.3, 3.2 Hz, 1H), 1.15 (dqd, J = 24.7, 12.2, 3.7 Hz, 2H). 13C NMR (126 MHz, CD3OD) δ 143.14, 137.67, 133.70, 131.73, 130.36, 130.33, 130.03, 129.28, 127.36, 126.84, 126.60, 125.27, 113.19, 59.65, 58.14, 53.26, 52.97, 48.19, 48.15, 48.12, 48.06, 47.95, 47.89, 47.78, 47.72, 47.61, 47.44, 47.32, 47.27, 47.10, 35.94, 35.86, 29.45, 20.03. HRMS (ESI): calcd. For C21H25F3N2O2S [M + H]+ 427.1662, found 427.1660.
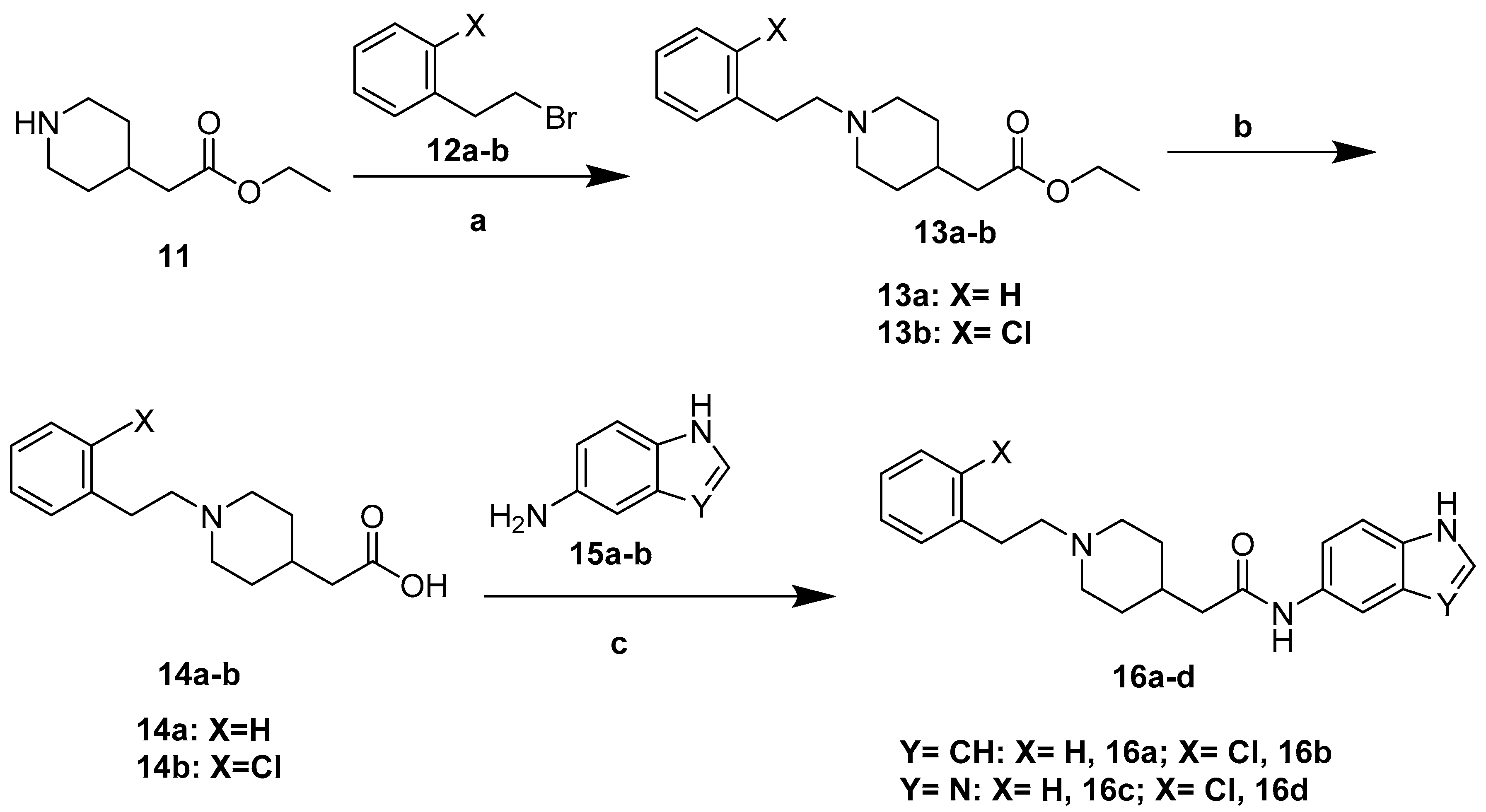
3.7. General Procedure for the Synthesis of 16a–d
Ethyl 2-piperidin-4-ylacetate 11 (1 g, 5.84 mmol, 1.0 equiv.) and substituted (2-romoethyl)benzene derivatives (12a–b) (7.01 mmol, 1.2 equiv.) were dissolved in 20 mL acetone. Then, anhydrous K2CO3 (11.68 mmol, 2 equiv.) and catalytic amount KI were added. The reaction mixture was refluxed for 4 h. After completion of the reaction, acetone was concentrated, and the residue was dissolved in water (60 mL) and extracted with ethyl acetate (60 × 3 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuum. The obtained oil was used in further synthesis without purification yielded compounds 13a–b (Yields were 67% and 72%). Then, 4 mol/L KOH (2.5 equiv.) was added to the solution of compounds 13a–b in C2H5OH: H2O = 5:1(6 mL). The reaction mixture was stirred at room temperature for 7 h. After completion of the reaction, the reaction mixture was evaporated to dryness after neutralization with dilute hydrochloric acid solution. Poured into ethyl acetate to deposit the solid, after cooling off, the mixture was filtered and washed with cold ethyl acetate to give compounds 14a–d.
Finally, intermediates (14a–b) (1.2 equiv.), PyBOP (1.2 equiv.) and DIPEA (1.5 equiv.) were added to 6 mL DMF and stirred at room temperature for 20 min. Then, intermediates (15a–b) (1.0 equiv.) was added and stirred at room temperature for 4 h. After completion of the reaction, the reaction mixture was quenched with saturated NaCl solution. The aqueous phase was extracted with DCM. The DCM layer was combined and washed with brine solution. The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. After concentration, the crude product was purified by silica gel column chromatograph using a methanol in dichloromethane gradient (DCM:methanol = 60:1–5:1) yielded compounds 16a–d.
N-(1H-Indol-5-yl)-2-(1-phenethylpiperidin-4-yl)acetamide (16a). 2-(1-Phenethylpiperidin-4-yl) acetic acid (140 mg, 0.57 mmol), 1H-Indol-5-amine (63 mg, 0.48 mmol), PyBOP (295 mg, 0.57 mmol), DIPEA (93 mg, 0.72 mmol), DMF (6 mL). Yellow solid, m.p.: 165–167 °C, yield: 80.70%, 1H NMR (400 MHz, CD3OD) δ 7.74 (d, J = 2.0 Hz, 1H), 7.28 (tt, J = 13.1, 6.8 Hz, 6H), 7.20 (d, J = 3.1 Hz, 1H), 7.15 (dd, J = 8.6, 2.0 Hz, 1H), 6.37 (d, J = 3.1 Hz, 1H), 3.60 (d, J = 14.9 Hz, 2H), 3.26 (d, J = 5.0 Hz, 1H), 3.08–3.01 (m, 3H), 2.39 (d, J = 7.0 Hz, 2H), 2.21–2.13 (m, 1H), 2.04 (d, J = 15.4 Hz, 2H), 1.74–1.60 (m, 2H), 1.27 (s, 2H). 13C NMR (151 MHz, CD3OD) δ 170.66, 136.38, 133.81, 129.79, 128.59, 128.42, 127.97, 126.86, 125.29, 115.83, 115.76, 112.47, 110.75, 101.07, 72.24, 70.09, 60.80, 57.69, 53.42, 52.37, 41.90, 31.68, 31.23, 29.38, 28.89, 22.34, 13.05. HRMS (ESI): calcd. For C23H27N3O [M + H]+ 362.2227, found 362.2242.
2-(1-(2-Chlorophenethyl)piperidin-4-yl)-N-(1H-indol-5-yl)acetamide (16b). 2-(1-(2-Chlorophenethyl)piperidin-4-yl)acetic acid (140 mg, 0.50 mmol), 1H-Indol-5-amine (55 mg, 0.42 mmol), PyBOP (260 mg, 0.50 mmol), DIPEA(80 mg, 0.62 mmol), DMF (6 mL). Yellow solid, m.p.: 172–173 °C, yield: 30.41%, 1H NMR (400 MHz, CD3OD) δ 7.75 (d, J = 1.9 Hz, 1H), 7.39–7.29 (m, 3H), 7.28–7.20 (m, 2H), 7.19 (d, J = 3.1 Hz, 1H), 7.16 (dd, J = 8.7, 2.0 Hz, 1H), 6.37 (d, J = 4.0 Hz, 1H), 3.45 (d, J = 12.3 Hz, 2H), 3.15–3.09 (m, 2H), 3.08–3.01 (m, 2H), 2.79 (t, J = 13.0 Hz, 2H), 2.36 (d, J = 7.1 Hz, 2H), 2.10 (s, 1H), 2.00–1.92 (m, 2H), 1.60 (q, J = 11.5 Hz, 2H). 13C NMR (151 MHz, CD3OD) δ 171.22, 135.56, 133.78, 133.57, 130.84, 129.85, 129.30, 128.28, 127.96, 127.17, 125.21, 115.80, 112.48, 110.73, 101.06, 56.91, 52.68, 42.55, 32.24, 29.95, 29.32, 29.01. HRMS (ESI): calcd. For C23H26ClN3O [M + H]+ 396.1837, found 396.1838.
N-(1H-Benzo[d]imidazol-5-yl)-2-(1-phenethylpiperidin-4-yl)acetamide (16c). 2-(1-phenethylpiperidin-4-yl)acetic acid (140 mg, 0.57 mmol), 1H-Benzo[d]imidazol-5-amine (64 mg, 0.48 mmol), PyBOP (295 mg, 0.57 mmol), DIPEA(93 mg, 0.72 mmol), DMF (6 mL). Red solid, m.p.: 167–168 °C, yield: 45.92%, 1H NMR (400 MHz, CD3OD) δ 8.09 (s, 1H), 8.03 (s, 1H), 7.52 (d, J = 8.7 Hz, 1H), 7.29–7.12 (m, 6H), 3.07 (d, J = 12.0 Hz, 2H), 2.84–2.78 (m, 2H), 2.64–2.58 (m, 2H), 2.32 (d, J = 7.2 Hz, 2H), 2.17 (t, J = 11.8 Hz, 2H), 1.93 (s, 1H), 1.81 (d, J = 12.9 Hz, 2H), 1.42 (q, J = 12.1, 10.5 Hz, 2H). 13C NMR (101 MHz, CD3OD) δ 171.84, 141.67, 139.67, 133.93, 128.33, 128.21, 128.20, 125.93, 116.32, 60.33, 53.19, 47.89, 43.30, 33.27, 32.51, 31.15. HRMS (ESI): calcd. For C22H26N4O [M + H]+ 363.2179, found 363.2197.
N-(1H-Benzo[d]imidazol-5-yl)-2-(1-(2-chlorophenethyl)piperidin-4-yl)acetamide (16d). 2-(1-(2-Chlorophenethyl)piperidin-4-yl)acetic acid (140 mg, 0.50 mmol), 1H-Benzo[d]imidazol-5-amine (55 mg, 0.42 mmol), PyBOP (259 mg, 0.50 mmol), DIPEA(80 mg, 0.62 mmol), DMF (6 mL). Red solid, m.p.: 176–177 °C, yield: 40.33%, 1H NMR (600 MHz, CDCl3) δ 7.95 (s, 1H), 7.70 (s, 1H), 7.66 (d, J = 8.1 Hz, 1H), 7.48 (d, J = 7.4 Hz, 1H), 7.35 (d, J = 7.8 Hz, 1H), 7.24 (t, J = 7.4 Hz, 1H), 7.19 (t, J = 7.3 Hz, 1H), 6.90 (d, J = 8.1 Hz, 1H), 3.62 (s, 2H), 3.57 (d, J = 11.7 Hz, 1H), 3.37 (d, J = 6.5 Hz, 2H), 2.95 (d, J = 11.2 Hz, 2H), 2.10 (t, J = 11.3 Hz, 2H), 1.75 (d, J = 12.4 Hz, 2H), 1.66 (s, 1H), 1.60 (s, 1H), 1.39 (q, J = 11.5 Hz, 2H). 13C NMR (151 MHz, CD3OD) δ 171.90, 141.65, 137.34, 134.98, 133.95, 133.58, 130.73, 129.20, 127.73, 126.97, 121.27, 116.28, 58.31, 58.27, 53.17, 53.06, 50.62, 43.39, 40.15, 33.39, 32.59, 31.29, 31.15, 30.24. HRMS (ESI): calcd. For C22H25ClN4O [M + H]+ 397.1790, found 397.1797.
3.8. AChE and BChE Inhibition Assay
The inhibitory activity of the target compound against AChE (from Electrophorus electricus (eeAChE), Sigma-Aldrich, Munich, Germany) and horse serum BChE (eqBChE, Sigma-Aldrich, Munich, Germany) were measured by Ellman’s method [
32]. In 96-well plates, a mixture of phosphate buffer (0.1 M, pH 8.0, 2 mL), 5,5′-dithiobis-2-nitrobenzoic acid (DTNB, 60 μL), acetylcholinesterase or butyrylcholinesterase (20 μL, 5 IU/mL) and different concentrations of the compounds solution (30 μL) was pre-incubated for 5 min and then substrates (acetylthiocholine iodide or butyrylthiocholine iodide, 20 μL) were added.
Changes in absorbance were measured at 412 nm by using microplate reader (Thermo, Varioskan Flash 3001, Thermo Fisher Scientific, Agawam, MA, USA). The measurement of each concentration for each compound was detected in triplicate. GraphPad Prism 6.0 (GraphPad Software, Inc., La Jolla, CA, USA) was used for data processing. The inhibition curve was fitted by plotting the logarithm of the concentration of the tested compounds with the percentage of enzyme activity (reference to 100%). The (IC50) value was calculated according to the inhibition curve and the data were shown in the layout of mean ± SEM by GraphPad Prism 6.0.
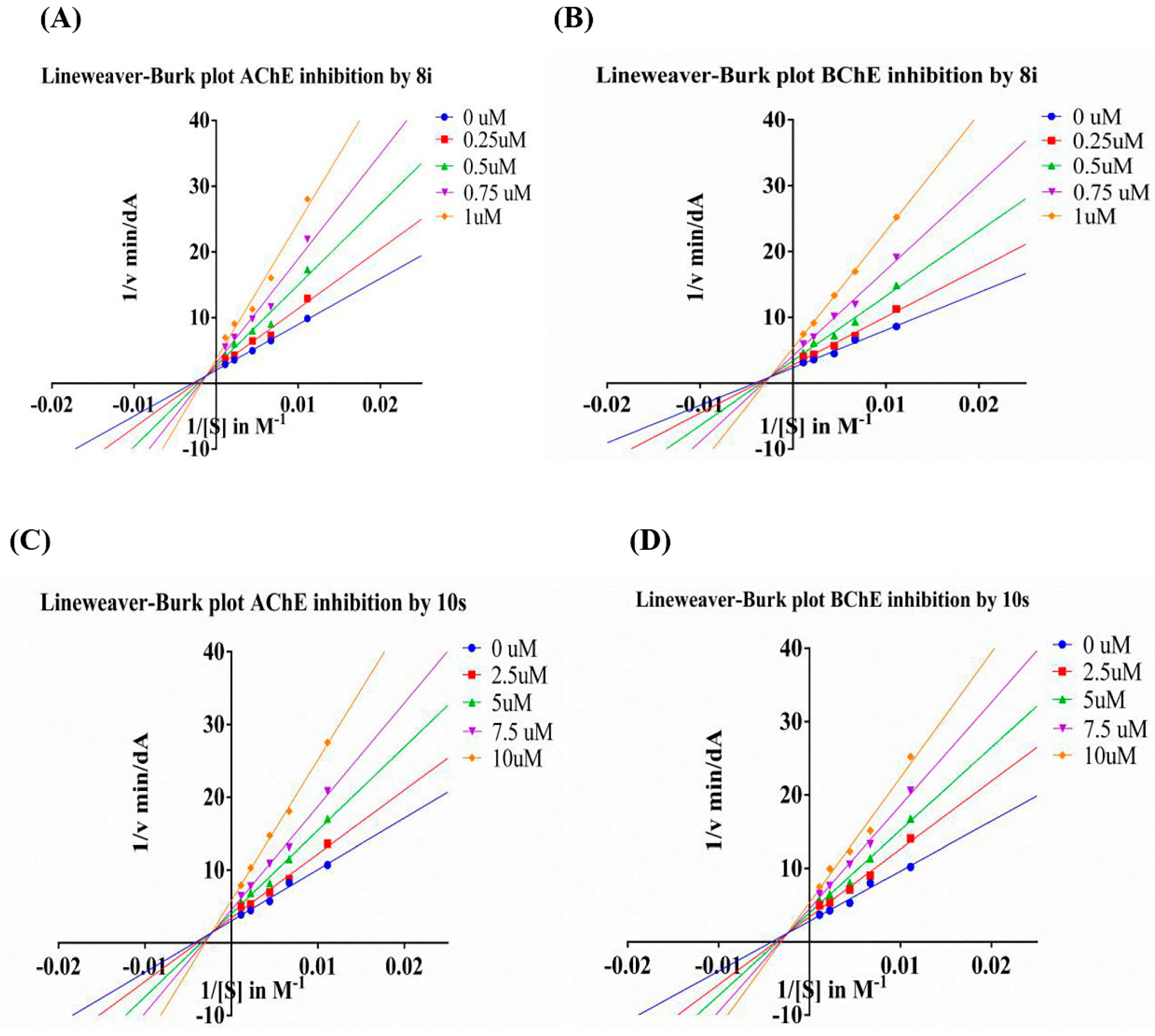
3.9. Kinetics of AChE and BChE Inhibition
Kinetic studies were performed in the same manner as the determination of ChEs inhibition, with substrate (ATC/BTC) concentrations of 90, 150, 226, 452 and 904 μM. The concentration of compound 8i was set to 0, 0.25, 0.5, 0.75, 1 μM, and the concentration of compound 10s was set to 1, 2.5, 5, 7.5, 10 μM. The enzymatic reaction was extended to 7 min for eeAChE and eqBChE before the determination of the absorption. The Vmax and Km values of Michaelis-Menten kinetics were calculated by nonlinear regression from the substrate-velocity curves using GraphPad Prism 6.0. Linear regression was used to fit the Lineweaver–Burk plot.
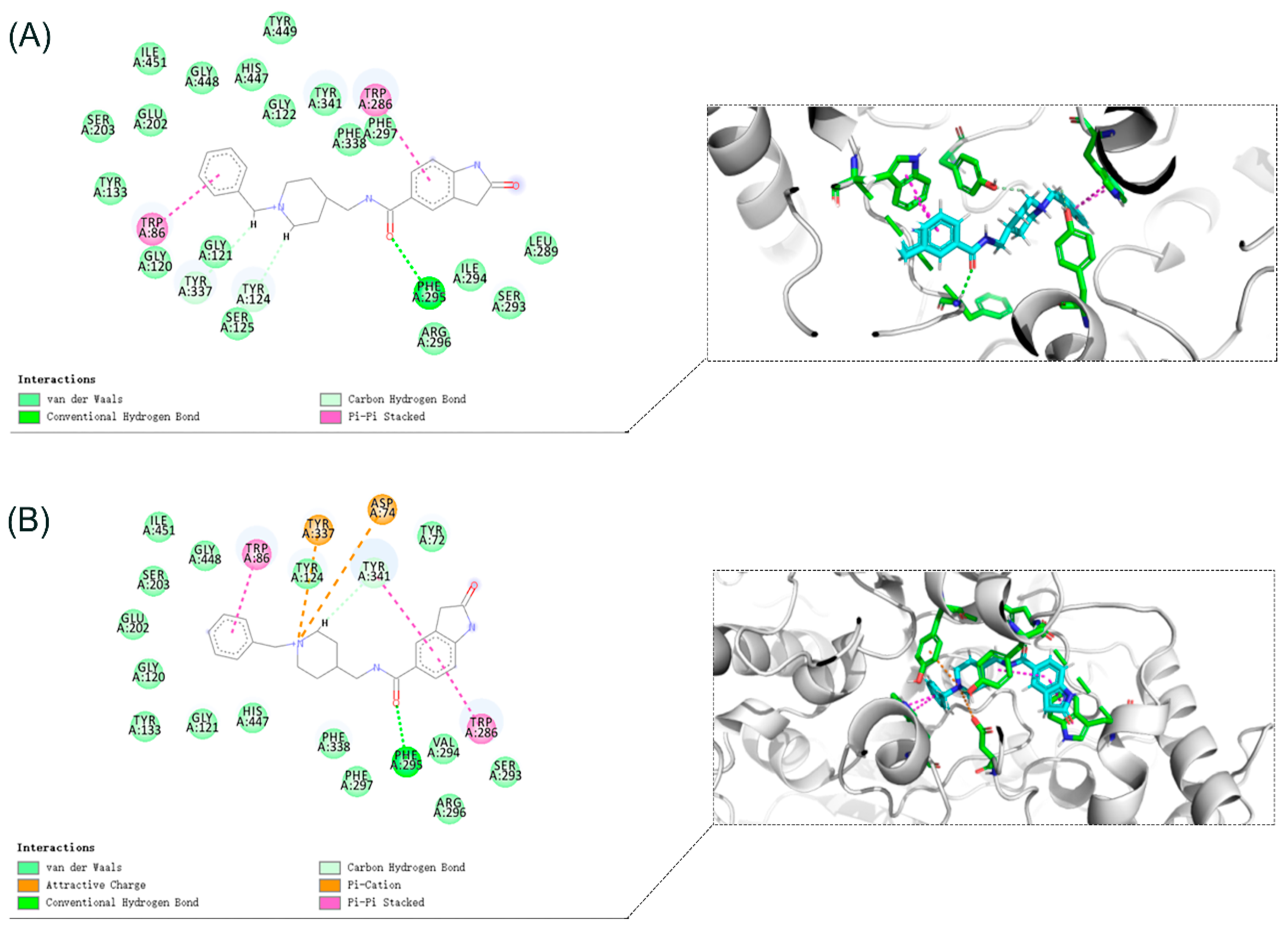
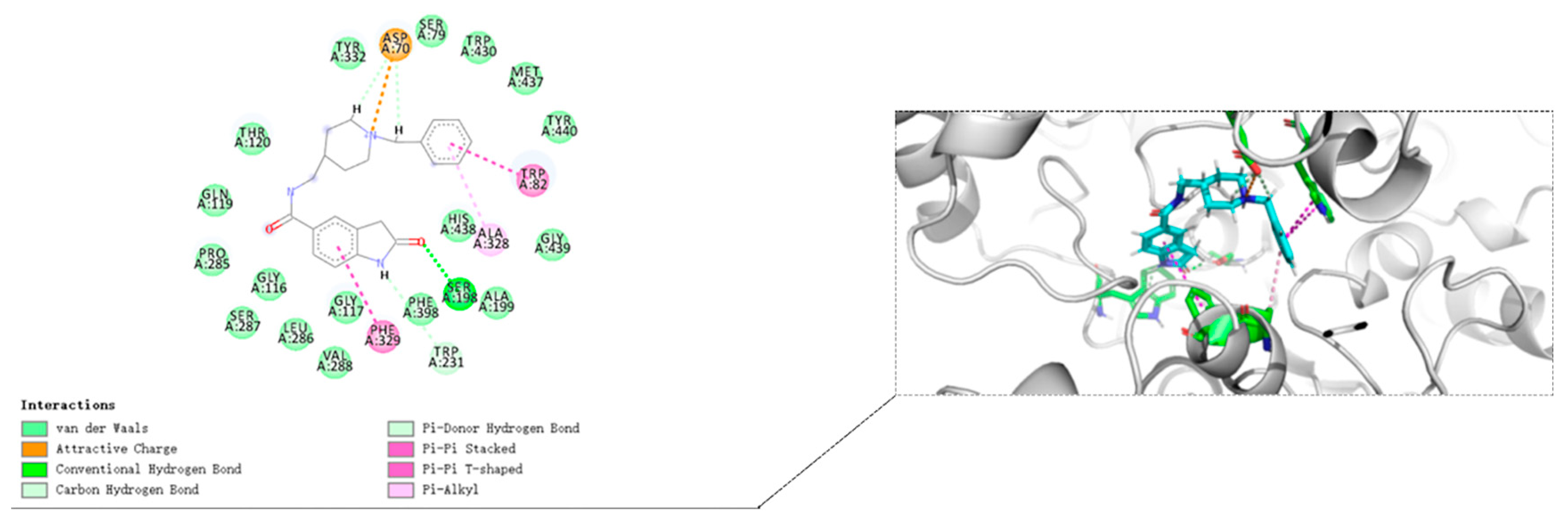
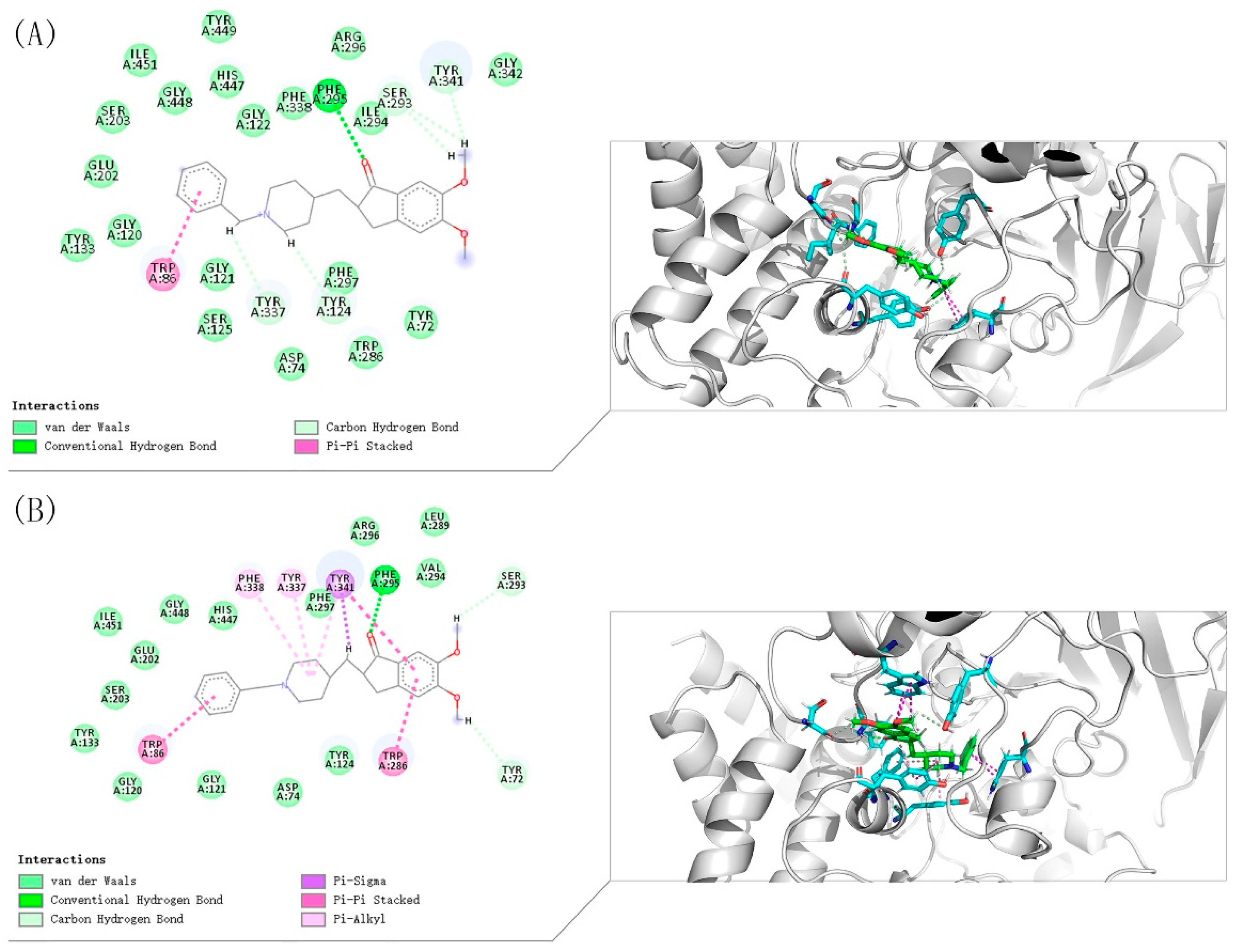
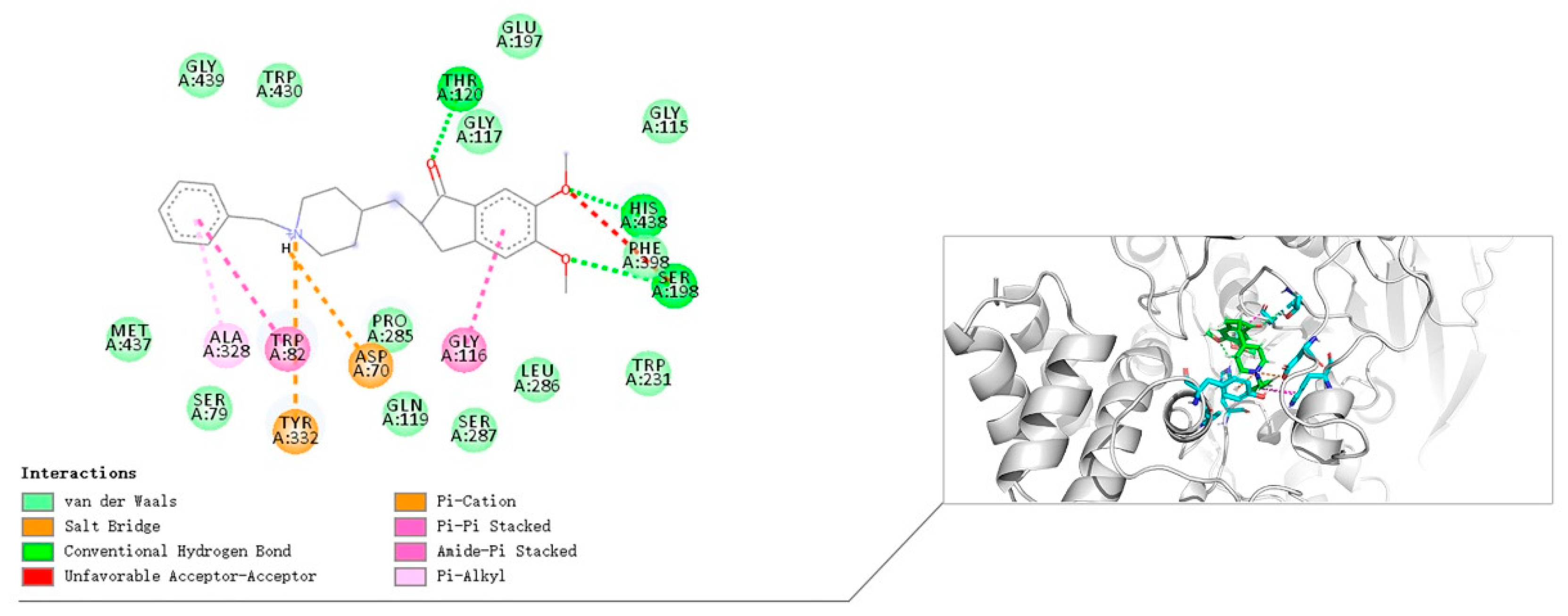
3.10. Molecular Docking Study
The crystal structures of eeAChE (PDB:1C2B) [
36], huAChE (PDB ID: 4EY7) [
32] and huBChE (PDB ID: 4TPK) [
37] were obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB). The Discovery Studio software 2016 (DS 2016, BIOVIA, San Diego, CA, USA) was used to study the docking of compound
8i. The three protein structures are pretreated (i.e., protonated, removed water, added Miss sidechains, etc.) by the “prepare protein” module in DS to provide the structures suitable for docking. The “prepare ligand” module in DS is used to test the structural preparation of the compound. The native ligand in the crystal structure was used to define the binding site. The binding site was defined as the site sphere ((in 10 Å radius) around the original ligand in the co-crystal structures. The docking program CDOCKER encoded in DS 2016 was applied to identify the potential binding of compound
8i to eeAChE, huAChE, and huBChE. Other CDOCKER parameters were set to default values. Compound
8i was chosen for molecular modeling as the most active compounds in the series (
Table 1). Compound
8i produced 10 poses to eeAChE, huAChE, and huBChE. These postures were visually examined, and the most appropriate docking pose was selected according to the scores and interactions with key residues of the eeAChE, huAChE, and huBChE active sites.
3.11. Cell Studies in Vitro
Pheochromocytoma-derived cell line (PC12 cells) were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS at 37 °C in a humidified atmosphere containing 5% CO
2. To carry out the experiment, cells (6 × 10
3 cells/well) were seeded in 96-well plate in complete medium. After 24 h, the culture medium was removed and the cells were exposed to increasing concentrations of compounds
6b,
6c,
6d,
8i,
10s,
10v or Tacrine (10, 20, 30 and 50 μM) in DMEM for further 24 h. Cell survival was measured by 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) assay [
37].















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

























