Bioassay-Guided Isolation and HPLC Quantification of Antiproliferative Metabolites from Stahlianthus thorelii

 ,
,  ,
,
Abstract
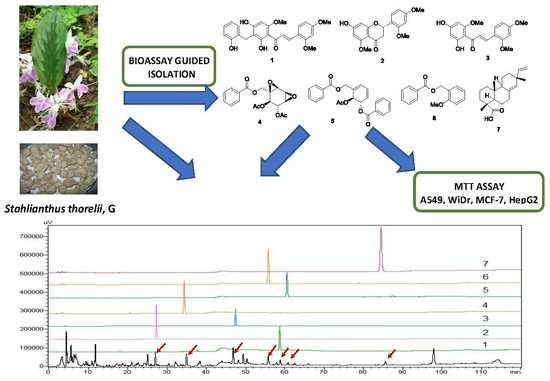
:
1. Introduction
2. Results and Discussion
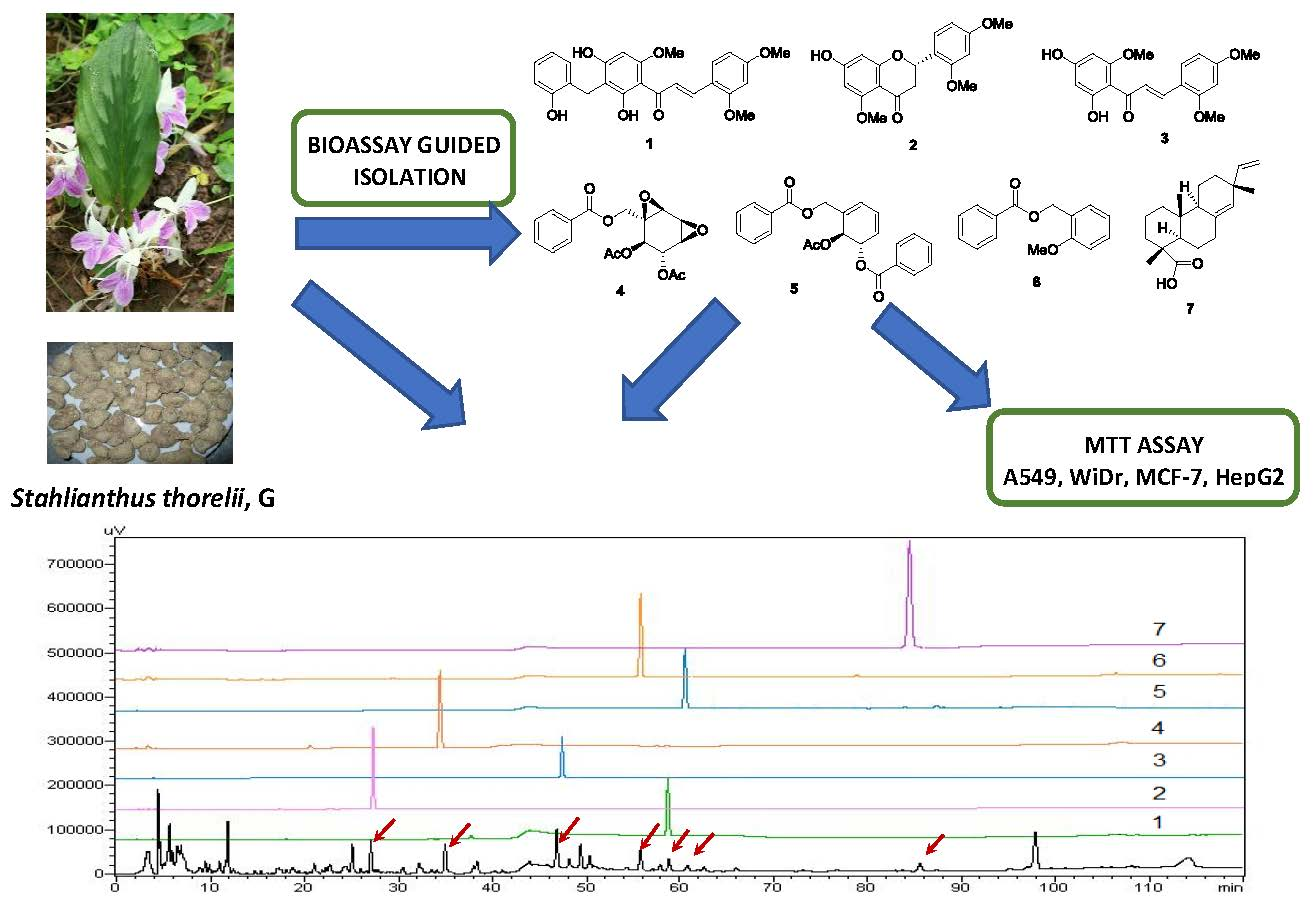
2.1. Bioactivity-Guided Isolation of Chemical Constituents
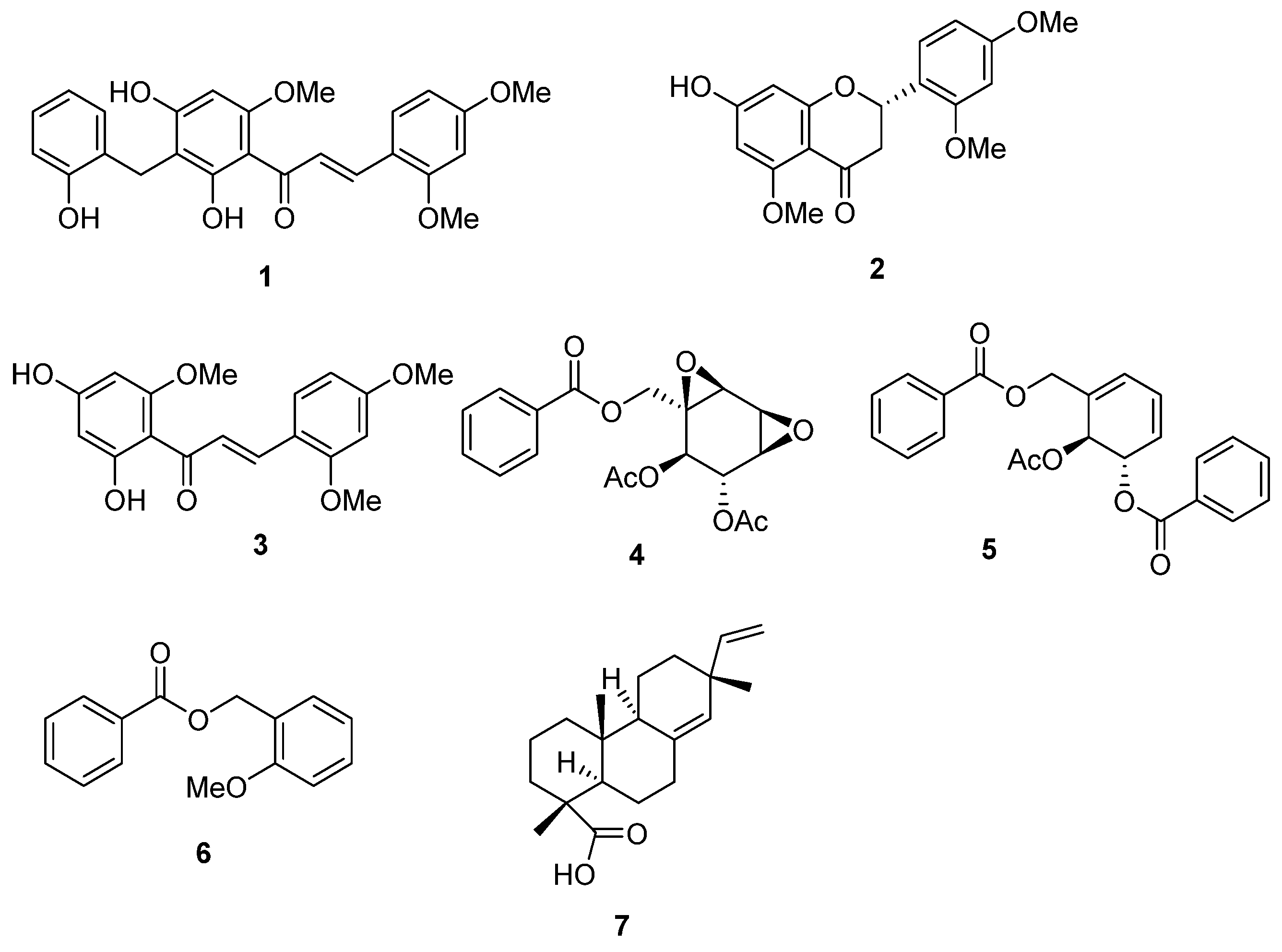
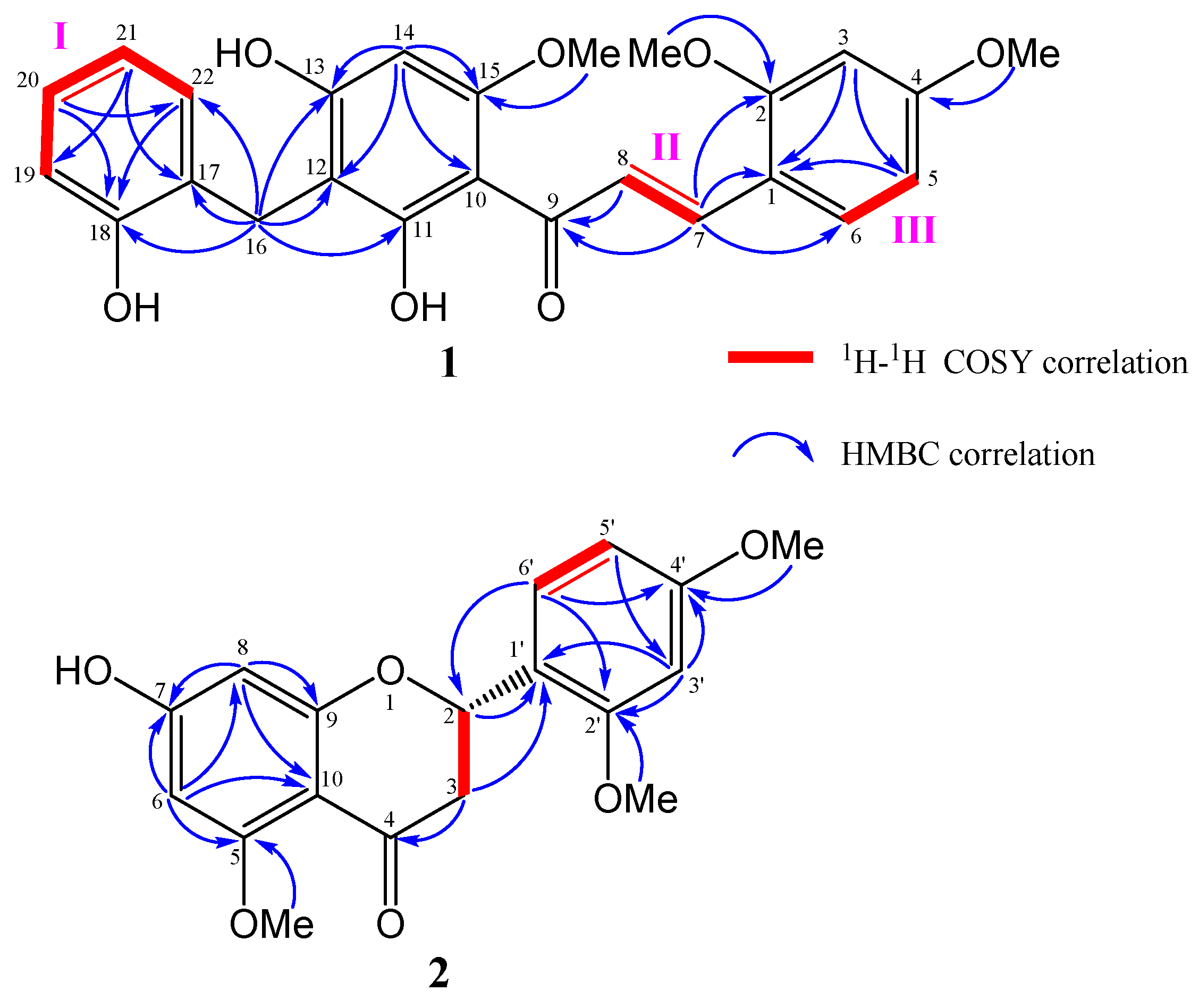
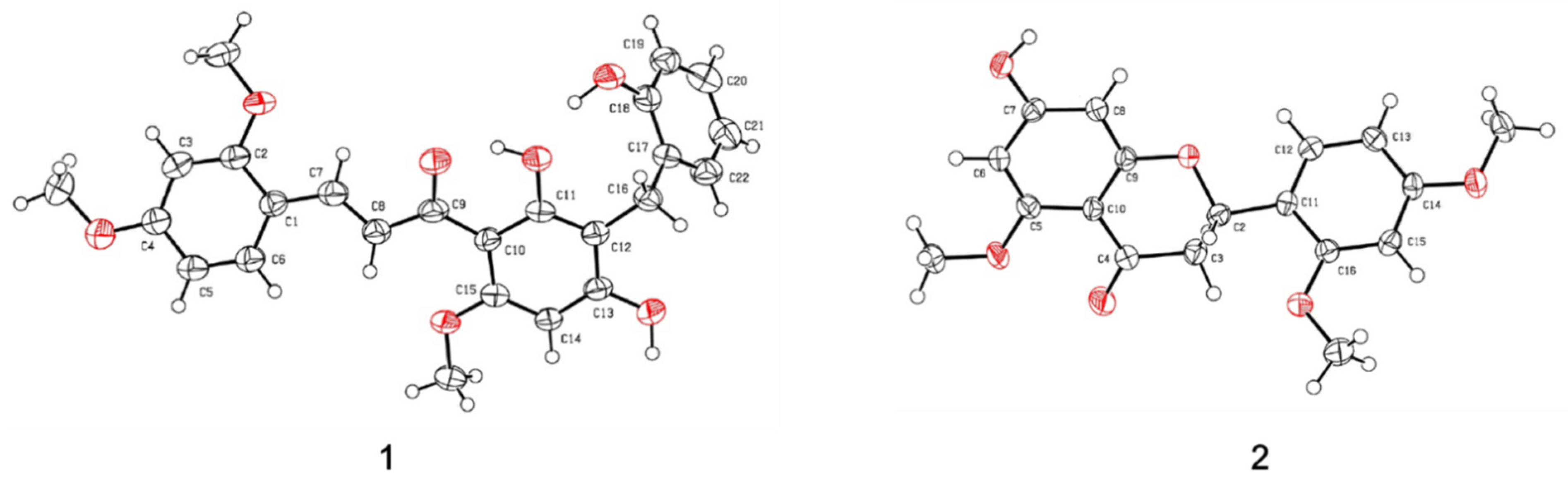
2.2. Structural Determination
2.3. Evaluation of the Anticancer Potential of Isolated Compounds
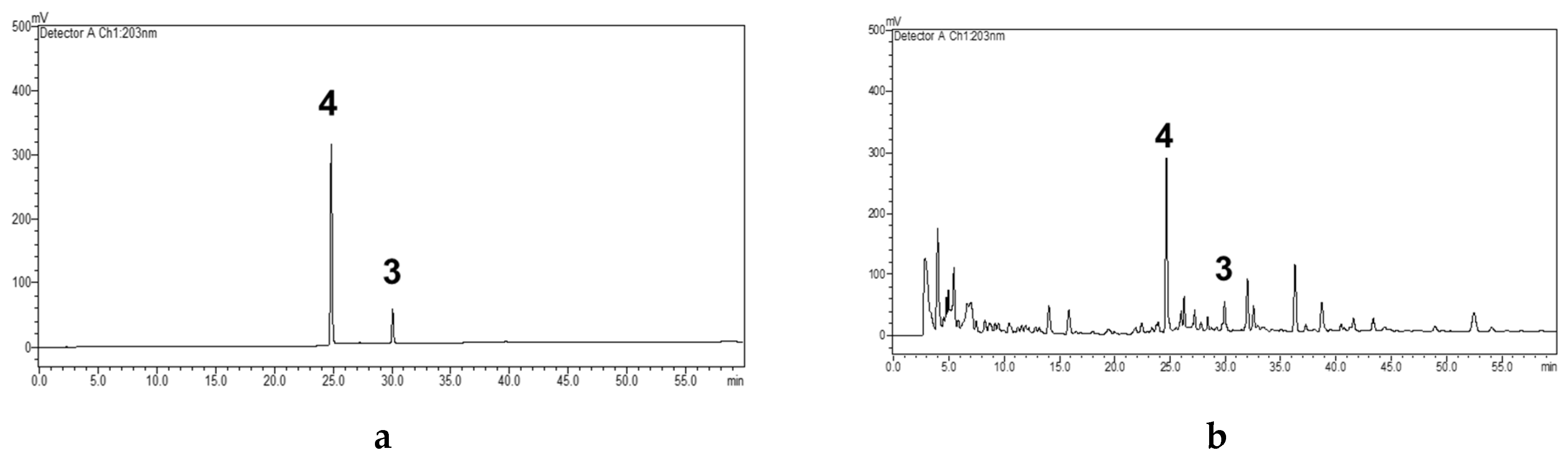
2.4. Quantitative Analysis of Compounds 3 and 4
2.5. Validation of the HPLC-UV Analysis Method
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. X-ray Crystallographic Analysis
3.5. Colorimetric MTT In Vitro Assay
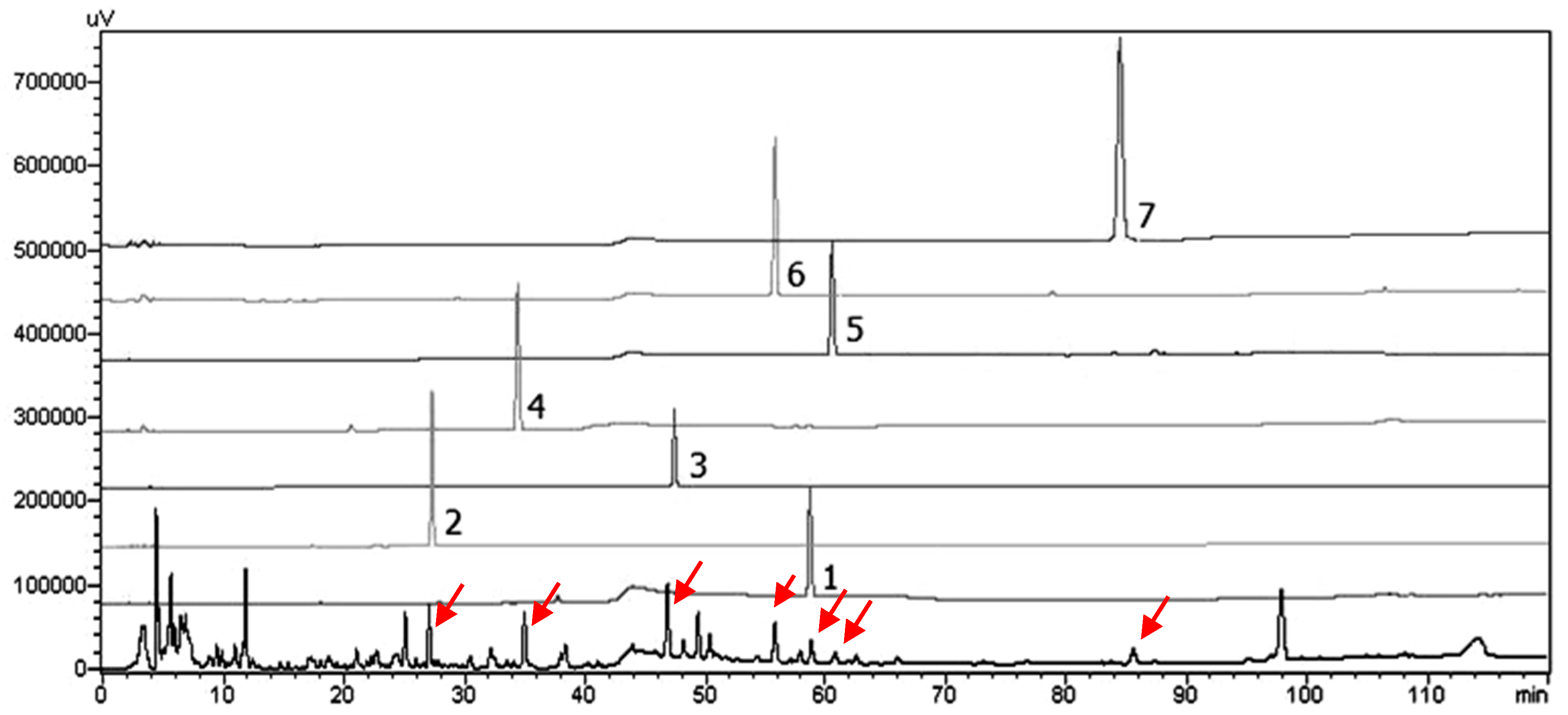
3.6. HPLC-SPD Separation Profile for Analysis of Seven Isolates from S. thorelii
3.7. Quantitation of Bioactive Compound 3 and Major Yielded Compound 4
3.7.1. Validation of the HPLC-UV Method
3.7.2. Preparation of Calibration Standards
3.7.3. Preparation of Validated Extracted Samples
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| A549 | human lung adenocarcinoma |
| CAN | acetonitrile |
| ATCC | American type culture collection |
| CDCl3 | chloroform |
| DMSO | dimethyl sulfoxide |
| HepG2 | human liver adenocarcinoma |
| HPLC | high performance liquid chromatography |
| HR-ESI-MS | high resolution electrospray ionization mass spectrometry |
| IC50 | 50% inhibitory concentration |
| MCF-7 | human breast adenocarcinoma |
| WiDr | human colon adenocarcinoma |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium |
| MEM | minimum essential medium |
| NMR | nuclear magnetic resonance |
| OD | optical density |
| PBS | phosphate buffered saline |
| TLC | thin-layer chromatography |
| MeOH | methanol |
| COSY | correlation spectroscopy |
| HMBC | heteronuclear multiple bonds correlation |
| NOESY | nuclear Overhauser effect spectroscopy |
| DEPT | distortionless enhancement by polarization transfer |
| FBS | fetal bovine serum |
| m/z | mass to charge ratio |
| Hz | hertz |
| CC | column chromatography |
| HSQC | homonuclear single quantum correlation |
| UV | ultraviolet spectroscopy |
| ICH | International Conference Harmonization |
| PVDF | polyvinylidene fluoride |
| RSD | relative standard deviation |
| IR | Infrared spectroscopy |
| HT29 | human colon adenocarcinoma |
| B164A5 | murine melanoma |
| JAK/STAT | Janus kinase/signal transducers and activators of transcription |
| EtOAc | ethyl acetate |
| CD | circular dichroism |
| SPD | photodiode array detector |
| ANOVA | analysis of variance |
References
- Lu, C.L.; Zhao, H.Y.; Jiang, J.G. Evaluation of multi-activities of 14 edible species from Zingiberaceae. Int. J. Food Sci. Nutr. 2013, 64, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Kirana, C.; Record, I.R.; McIntosh, G.H.; Jones, G.P. Screening for antitumor activity of 11 species of indonesian zingiberaceae using human MCF-7 and HT-29 cancer cells. Pharma. Biol. 2003, 41, 271–276. [Google Scholar] [CrossRef]
- Danciu, C.; Vlaia, L.; Fetea, F.; Hancianu, M.; Coricovac, D.E.; Ciurlea, S.A.; Şoica, C.M.; Marincu, I.; Vlaia, V.; Dehelean, C.A.; et al. Evaluation of phenolic profile, antioxidant and anticancer potential of two main representants of Zingiberaceae family against B164A5 murine melanoma cells. Biol. Res. 2015, 48, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Rao, K.V.; Samikkannu, T.; Dakshayani, K.B.; Zhang, X.; Sathaye, S.S.; Indap, M.A.; Nair, M.P. Chemopreventive potential of an ethyl acetate fraction from Curcuma longa is associated with up-regulation of p57kip2 and Rad9 in the PC3M prostate cancer cell line. Asian Pac. J. Cancer Prev. 2012, 13, 1031–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.L.; Liu, Q.; Peng, Y.B.; Qi, L.W.; Li, P. Steamed ginger (Zingiber officinale): Changed chemical profile and increased anticancer potential. Food Chem. 2011, 129, 1785–1792. [Google Scholar] [CrossRef]
- Park, G.H.; Park, J.H.; Song, H.M.; Eo, H.J.; Kim, M.K.; Lee, J.W.; Lee, M.H.; Cho, K.H.; Lee, J.R.; Cho, H.J.; et al. Anti-cancer activity of Ginger (Zingiber officinale) leaf through the expression of activating transcription factor 3 in human colorectal cancer cells. BMC Complement. Altern. Med. 2014, 14, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirana, C.; Record, I.R.; Jones, G.P.; McIntosh, G.H. Anticancer properties of panduratin A isolated from Boesenbergia pandurata (Zingiberaceae). J. Nat. Med. 2007, 61, 131–137. [Google Scholar] [CrossRef]
- Muangnoi, P.; Lu, M.; Lee, J.; Thepouyporn, A.; Mirzayans, R.; Le, X.C.; Weinfeld, M.; Changbumrung, S. Cytotoxicity, Apoptosis and DNA Damage Induced by Alpinia galangal Rhizome Extract. Planta Med. 2007, 73, 748–754. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sun, W.; Han, N.; Zou, Y.; Yin, D. Curcumin inhibits proliferation, migration, invasion and promotes apoptosis of retinoblastoma cell lines through modulation of miR-99a and JAK/STAT pathway. BMC Cancer 2018, 18, 1230. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.M.; Luo, J.G.; Wang, R.Z.; Wang, X.B.; Yang, M.H.; Luo, J.; Kong, L.Y. Involucratusins A-H: Unusual Cadinane Dimers from Stahlianthus involucratus with Multidrug Resistance Reversal Activity. Sci. Rep. 2016, 6, 29744. [Google Scholar] [CrossRef]
- Vo, V.C. Tu dien cay thuoc Viet Nam-Vietnamese Herbal Medicine Dictionary; Medical Publishing House: Ha Noi, Vietnam, 2012; pp. 769–770. [Google Scholar]
- Yu, J.G.; Chen, Y.H.; Fang, H.J. Studies on the medicinal plants of Chinese Zingiberaceae. Acta. Pharm. Sin. 1983, 18, 839–842. [Google Scholar]
- Li, Q.M.; Luo, J.G.; Zhang, Y.M.; Li, Z.R.; Wang, X.B.; Yang, M.H.; Luo, J.; Sun, H.B.; Chen, Y.J.; Kong, L.Y. Involucratustones A-C: Unprecedented sesquiterpene dimers containing multiple contiguous quaternary carbons from stahlianthus involucratus. Chemistry 2015, 21, 13206–13209. [Google Scholar] [CrossRef] [PubMed]
- Pingsusaen, P.; Kunanusorn, P.; Khonsung, P.; Chiranthanut, N.; Panthong, A.; Rujjanawate, C. Investigation of anti-inflammatory, antinociceptive and antipyretic activities of Stahlianthus involucratus rhizome ethanol extract. J. Ethnopharmacol. 2015, 162, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, G.R.; Parmar, V.S. Three new flavonoids in Prunus cerasus. Phytochemisty 1977, 16, 1317–1318. [Google Scholar] [CrossRef]
- Aponte, J.C.; Castillo, D.; Estevez, Y.; Gonzalez, G.; Arevalo, J.; Hammond, G.B.; Sauvain, M. In vitro and in vivo anti-Leishmania activity of polysubstituted synthetic chalcones. Bioorg. Med. Chem. Lett. 2010, 20, 100–103. [Google Scholar] [CrossRef]
- Makhuvele, R.; Foubert, K.; Apers, S.; Pieters, L.; Verschaeve, L.; Elgorashi, E. Antimutagenic constituents from Monanthotaxis caffra (Sond.) Verdc. J. Pharm. Pharmacol. 2018, 70, 976–984. [Google Scholar] [CrossRef] [Green Version]
- Montree Kodpinida, C.S. Chachanat Thebtaranonth, Yodhathai Thebtaranont, Structures of β-senepoxide, tingtanoxide, and their diene precursors. Constituents of Uvaria ferruginea. Tetrahedron Lett. 1983, 24, 2019–2022. [Google Scholar] [CrossRef]
- Awale, S.; Ueda, J.Y.; Athikomkulchai, S.; Abdelhamed, S.; Yokoyama, S.; Saiki, I.; Miyatake, R. Antiausterity agents from Uvaria dac and their preferential cytotoxic activity against human pancreatic cancer cell lines in a nutrient-deprived condition. J. Nat. Prod. 2012, 75, 1177–1183. [Google Scholar] [CrossRef]
- Muto, N.; Tomokuni, T.; Haramoto, M.; Tatemoto, H.; Nakanishi, T.; Inatomi, Y.; Murata, H.; Inada, A. Isolation of apoptosis- and differentiation-inducing substances toward human promyelocytic leukemia HL-60 cells from leaves of Juniperus taxifolia. Biosci. Biotechnol. Biochem. 2008, 72, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Awale, S.; Tawila, A.M.; Dibwe, D.F.; Ueda, J.Y.; Sun, S.; Athikomkulchai, S.; Balachandran, C.; Saiki, I.; Matsumo, K.; Esumi, H. Highly oxygenated antiausterity agents from the leaves of Uvaria dac. Bioorg. Med. Chem. Lett. 2017, 27, 1967. [Google Scholar] [CrossRef]
- Lekphrom, R.; Kanokmedhakul, K.; Schevenels, F.; Kanokmedhakul, S. Antimalarial polyoxygenated cyclohexene derivatives from the roots of Uvaria cherrevensis. Fitoterapia 2018, 127, 420. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–7 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | IC50 (µg/mL) | |||
|---|---|---|---|---|
| A549 | MCF-7 | WiDr | HepG2 | |
| SEA | 72.83 ± 2.42 | 69.08 ± 1.24 | 46.81 ± 5.49 | 43.48 ± 4.84 |
| SF3 | >100 | >100 | 43.42 ± 0.55 | >100 |
| SF7 | 10.52 ± 0.51 | 7.89 ± 0.59 | 25.49 ± 0.87 | 76.38 ± 0.47 |
| SF9 | 13.12 ± 0.62 | 12.07 ± 1.61 | 20.04 ± 2.25 | 46.15 ± 1.42 |
| SF3.5 | 51.67 ± 4.64 | 52.09 ± 1.31 | 25.5 ± 1.15 | 43.87 ± 2.71 |
| SF3.6 | 25.18 ± 2.88 | 64.16 ± 5.08 | 55.69 ± 1.36 | 21.29 ± 0.72 |
| No | 1 | No | 2 | ||
|---|---|---|---|---|---|
| δH | δC | δH | δC | ||
| 1 | 118.1 | 1 | |||
| 2 | 161.2 | 2 | 5.55 (dd, 12.5, 2.5) | 73.0 | |
| 3 | 6.64 (d, 2.0) | 99.1 | 3 | 2.95 (dd, 16.5, 12.5) 2.67 (dd, 16.5, 2.5) | 44.0 |
| 4 | 164.1 | 4 | 187.9 | ||
| 5 | 6.60 (dd, 8.5, 2.0) | 107.0 | 5 | 164.5 | |
| 6 | 7.65 (d, 8.5) | 131.2 | 6 | 5.95 (d, 2.5) | 95.6 |
| 7 | 8.02 (d, 15.5) | 126.0 | 7 | 164.4 | |
| 8 | 8.09 (d, 15.5) | 138.5 | 8 | 6.06 (d, 2.5) | 93.3 |
| 9 | 193.5 | 9 | 162.2 | ||
| 10 | 105.9 | 10 | 104.3 | ||
| 11 | 166.4 | 1′ | 119.1 | ||
| 12 | 108.2 | 2′ | 160.7 | ||
| 13 | 164.4 | 3′ | 6.61 (d, 2.0) | 105.0 | |
| 14 | 6.20 (s) | 92.5 | 4′ | 157.3 | |
| 15 | 162.6 | 5′ | 6.57 (dd, 8.0, 2.0) | 98.5 | |
| 16 | 3.89 (s) | 23.2 | 6’ | 7.38 (d, 8.0) | 127.7 |
| 17 | 128.2 | 5-OMe | 3.73 (s) | 55.7 | |
| 18 | 155.6 | 2′-OMe | 3.79 (s) | 55.3 | |
| 19 | 6.80 (dd, 8.0, 2.0) | 116.2 | 4′-OMe | 3.78 (s) | 55.6 |
| 20 | 6.98 (ddd, 8.0, 7.5, 2.0) | 127.7 | |||
| 21 | 6.71 (ddd, 8.0, 7.5, 2.0) | 120.3 | |||
| 22 | 7.27 (dd, 7.5, 2.0) | 131.5 | |||
| 2-OMe | 3.95 (s) | 56.1 | |||
| 4-OMe | 3.92 (s) | 56.2 | |||
| 15-OMe | 3.87 (s) | 55.9 | |||
| Compound | IC50 (µM) | |||
|---|---|---|---|---|
| A549 | MCF-7 | WiDr | HepG2 | |
| 1 | 19.93 ± 0.59 b | >200 | 39.05 ± 1.61 b | 9.05 ± 1.47 b |
| 2 | >200 | >200 | >200 | >200 |
| 3 | >200 | >200 | 72.09 ± 1.15 c | 11.25 ± 0.41 b |
| 4 | >200 | >200 | >200 | 77.74 ± 5.28 c |
| 5 | 94.54 ± 0.45 c | >200 | >200 | >200 |
| 6 | >200 | >200 | >200 | 153.25 ± 5.74 d |
| Mitomycin c | 0.32 ± 0.03 a | 0.5 ± 0.03 | 0.56 ± 0.03 a | 0.29 ± 0.03 a |
| Place to Collect | Collected Date | 4 (%) | 3 (%) |
|---|---|---|---|
| An Giang province | 01/12/2018 | 0.109 | 0.013 |
| Ho Chi Minh city | 02/12/2018 | 0.106 | 0.012 |
| Dong Nai province | 02/12/2018 | 0.102 | 0.013 |
| Average | 0.106 | 0.013 |
| Validation Criteria | 4 | 3 | |
|---|---|---|---|
| Linearity | |||
| Regression equation | Y = 32638X | Y = 57440X | |
| Linear range (µg/mL) | 2.5–500 | 2.5–500 | |
| R2 (≤2) | 0.9956 | 0.9969 | |
| LOD | 0.05 µg/mL | 0.025 µg/mL | |
| LOQ | 0.17 µg/mL | 0.08 µg/mL | |
| System suitability | |||
| R2-tR (≤2) | 0.1443 | 0.0676 | |
| R2–Area (≤2) | 0.1358 | 1.2298 | |
| R2–T. plate (≤2) | 1.5610 | 1.1262 | |
| R2–Resolution (≤2) | 1.8971 | 1.7550 | |
| Repeatability (n = 6) | |||
| R2-tR (≤2) | 0.1469 | 0.1136 | |
| R2–Area (≤5.3) | 3.3516 | 2.5904 | |
| Accuracy | |||
| Level 1 (n = 3) | % recovery | 96.55 | 96.12 |
| R2 (≤5.3) | 2.74 | 5.15 | |
| Level 2 (n = 3) | % recovery | 95.8 | 87.50 |
| R2 (≤5.3) | 3.00 | 4.25 | |
| Level 3 (n = 3) | % recovery | 94.05 | 97.50 |
| R2 (≤5.3) | 2.58 | 4.01 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, N.-L.; Vo, T.-H.; Lin, Y.-C.; Liaw, C.-C.; Lin, Z.-H.; Chen, M.-C.; Kuo, Y.-H. Bioassay-Guided Isolation and HPLC Quantification of Antiproliferative Metabolites from Stahlianthus thorelii. Molecules 2020, 25, 551. https://doi.org/10.3390/molecules25030551
Nguyen N-L, Vo T-H, Lin Y-C, Liaw C-C, Lin Z-H, Chen M-C, Kuo Y-H. Bioassay-Guided Isolation and HPLC Quantification of Antiproliferative Metabolites from Stahlianthus thorelii. Molecules. 2020; 25(3):551. https://doi.org/10.3390/molecules25030551
Chicago/Turabian StyleNguyen, Nham-Linh, Thanh-Hoa Vo, Yu-Chi Lin, Chia-Ching Liaw, Zhi-Hu Lin, Mei-Chuan Chen, and Yao-Haur Kuo. 2020. "Bioassay-Guided Isolation and HPLC Quantification of Antiproliferative Metabolites from Stahlianthus thorelii" Molecules 25, no. 3: 551. https://doi.org/10.3390/molecules25030551
APA StyleNguyen, N. -L., Vo, T. -H., Lin, Y. -C., Liaw, C. -C., Lin, Z. -H., Chen, M. -C., & Kuo, Y. -H. (2020). Bioassay-Guided Isolation and HPLC Quantification of Antiproliferative Metabolites from Stahlianthus thorelii. Molecules, 25(3), 551. https://doi.org/10.3390/molecules25030551