
Recent Progress in Nitro-Promoted Direct Functionalization of Pyridones and Quinolones



Abstract
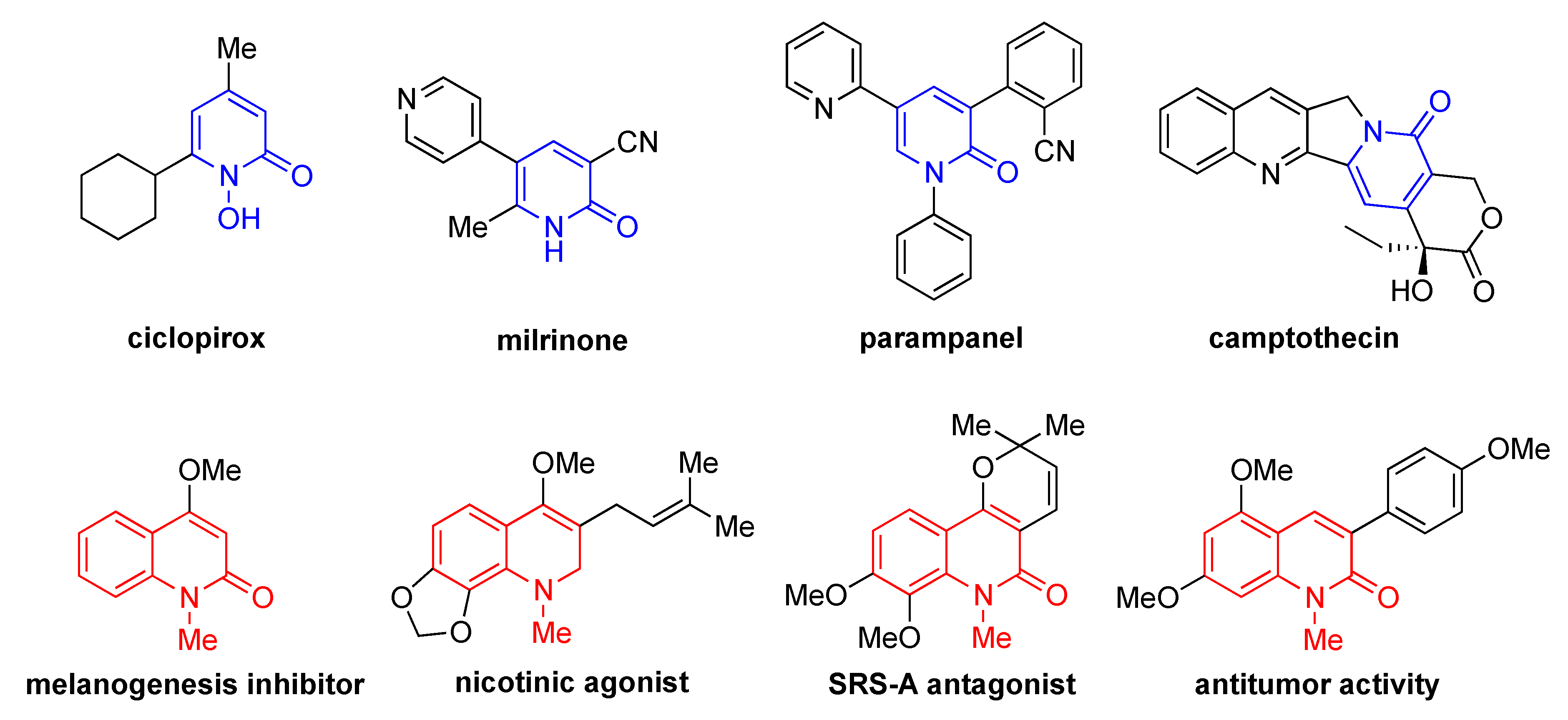
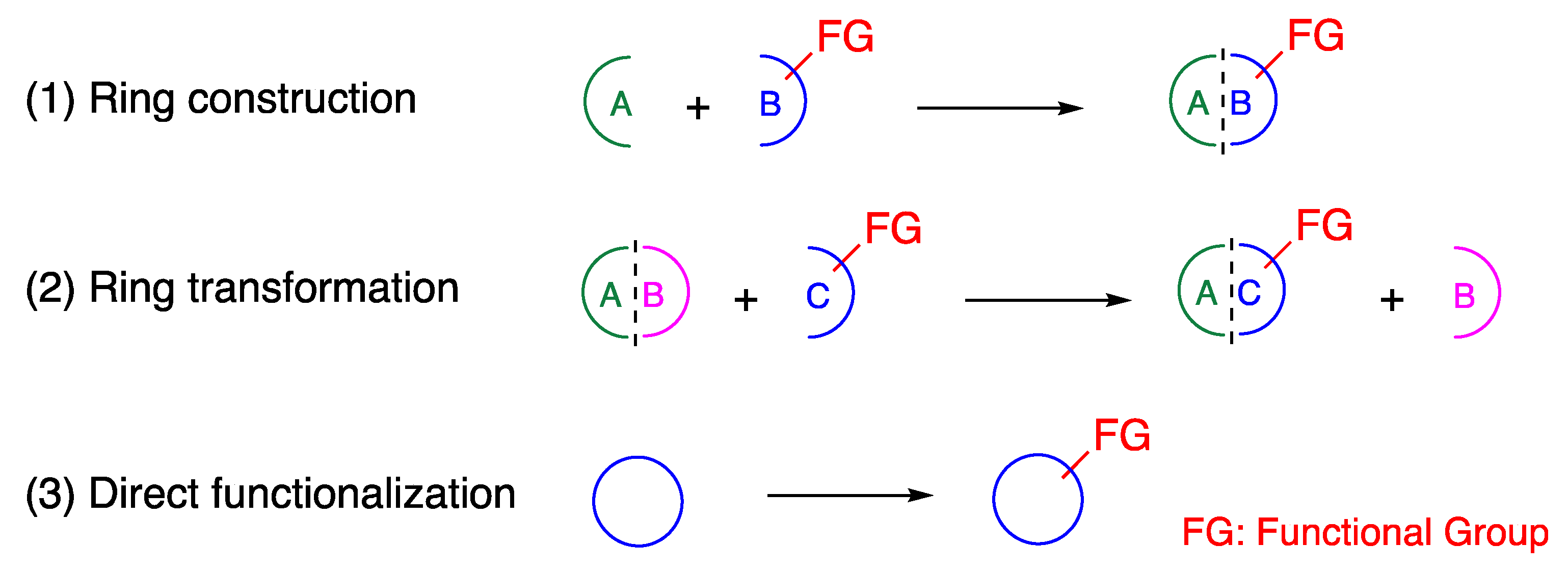
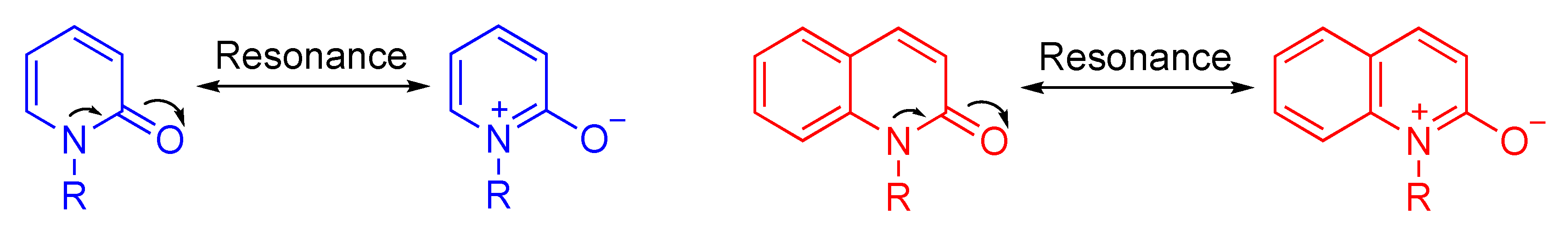
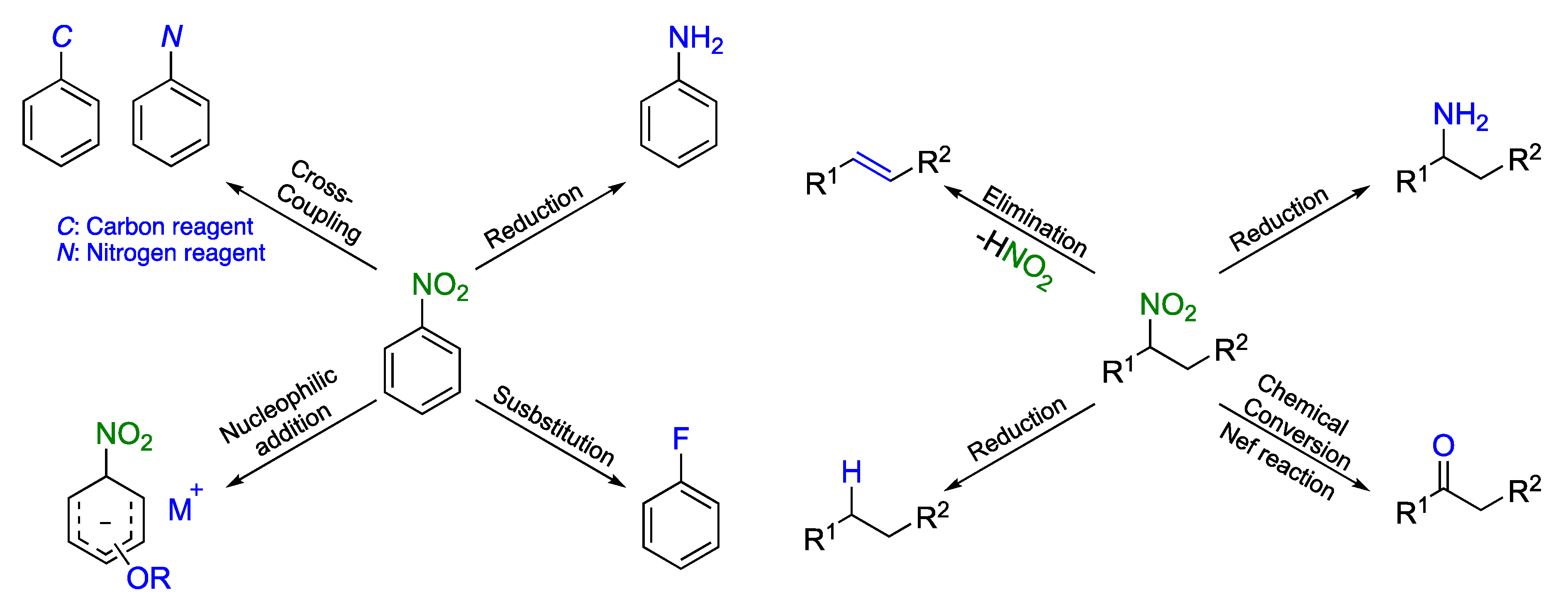
:1. Introduction
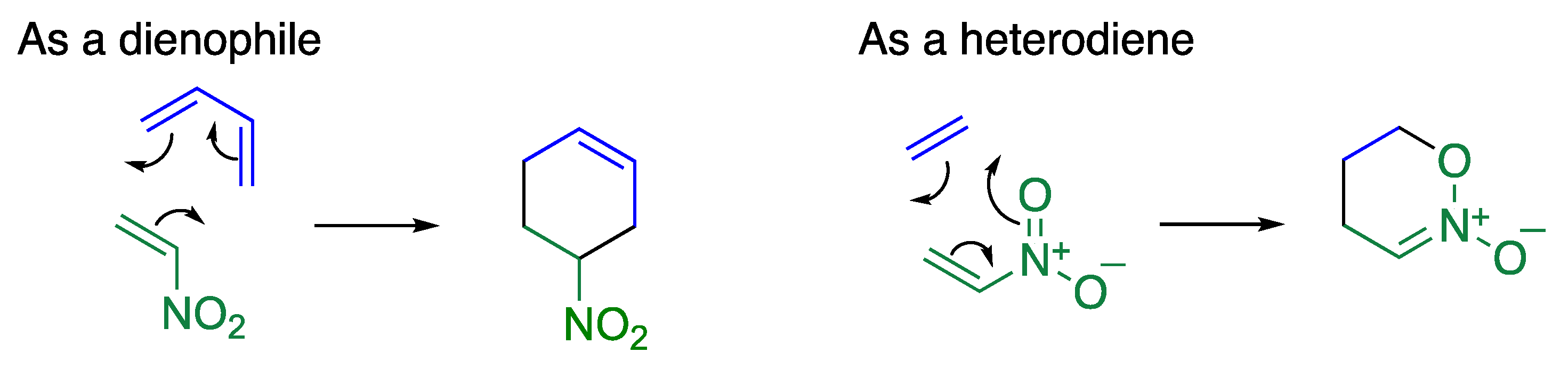
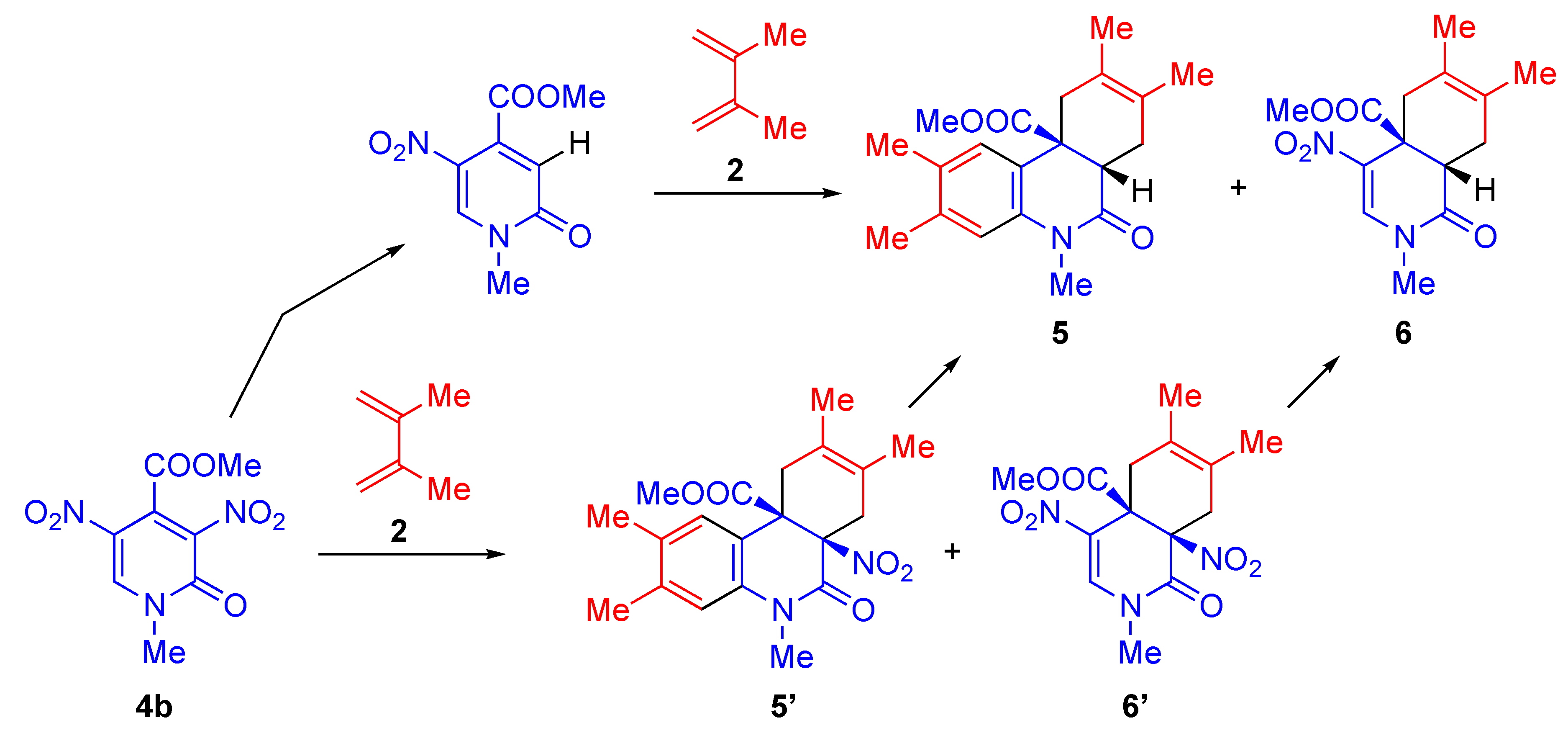
2. Cycloaddition of Nitropyridones
3. Cycloaddition of Nitroquinolones
4. Nitro-Promoted Cyclization of Pyridones via Nucleophilic Addition
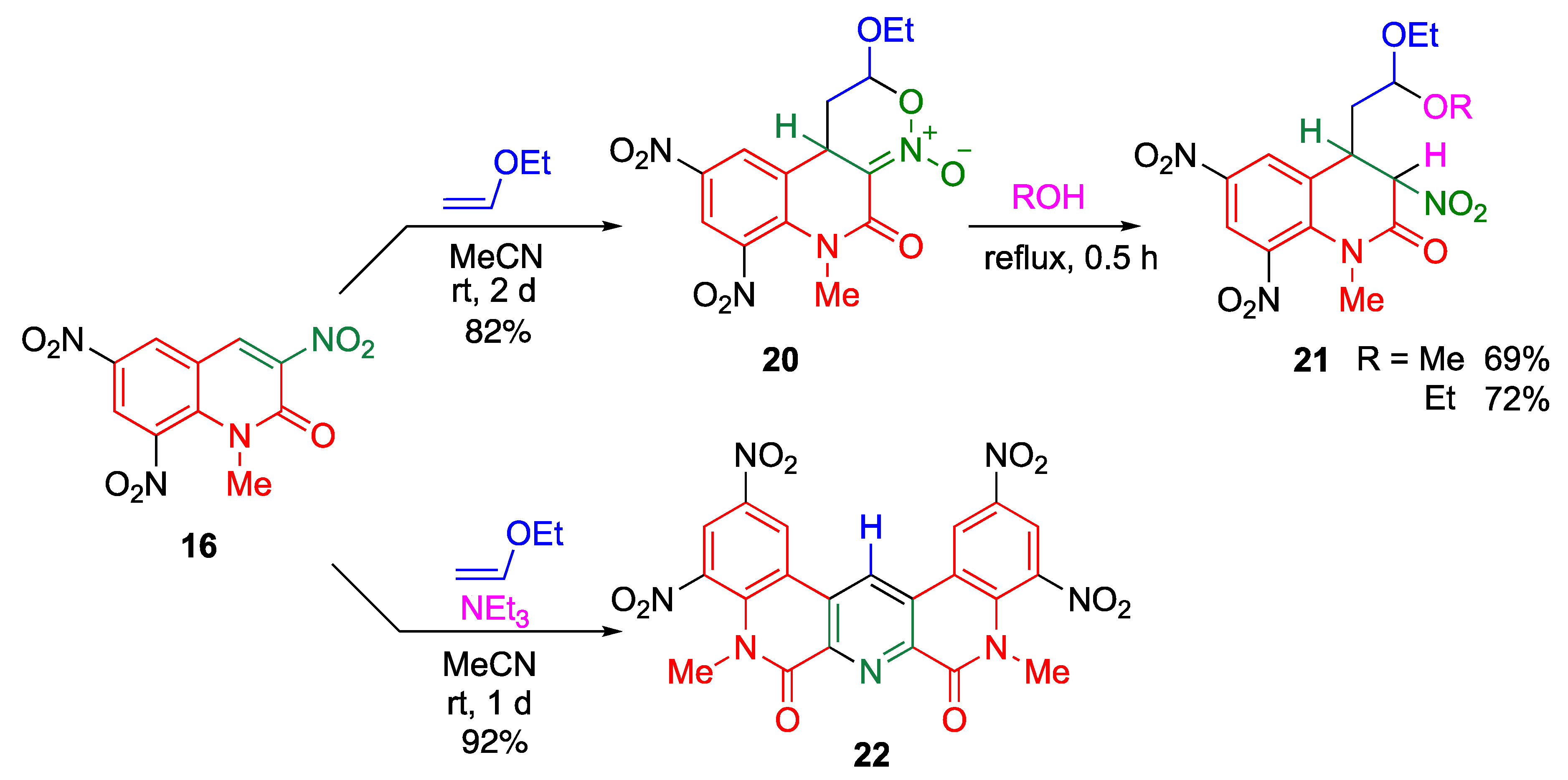
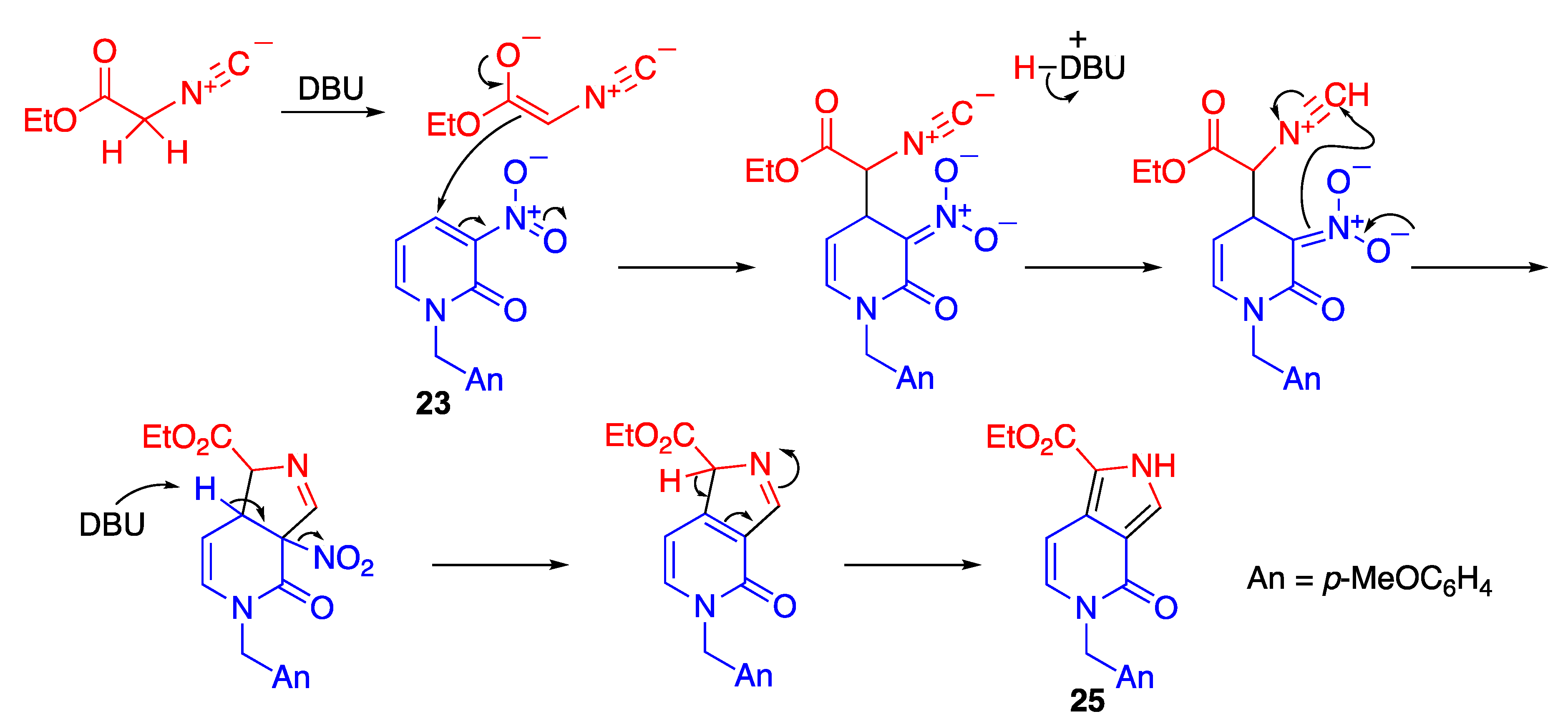
5. Nitro-Promoted Direct Functionalization of Quinolones
5.1. Direct C–C Bond Formation at the 4-Position via Cine-Substitution
5.1.1. cine-Substitution of Trinitroquinolone with 1,3-dicarbonyl Compounds
5.1.2. cine-Substitution of Trinitroquinolone with Nitroalkanes
5.1.3. cine-Substitution of Trinitroquinolone with Aldehyde, Ketones and Enamines
5.1.4. cine-Substitution of Trinitroquinolone with Phenoxides
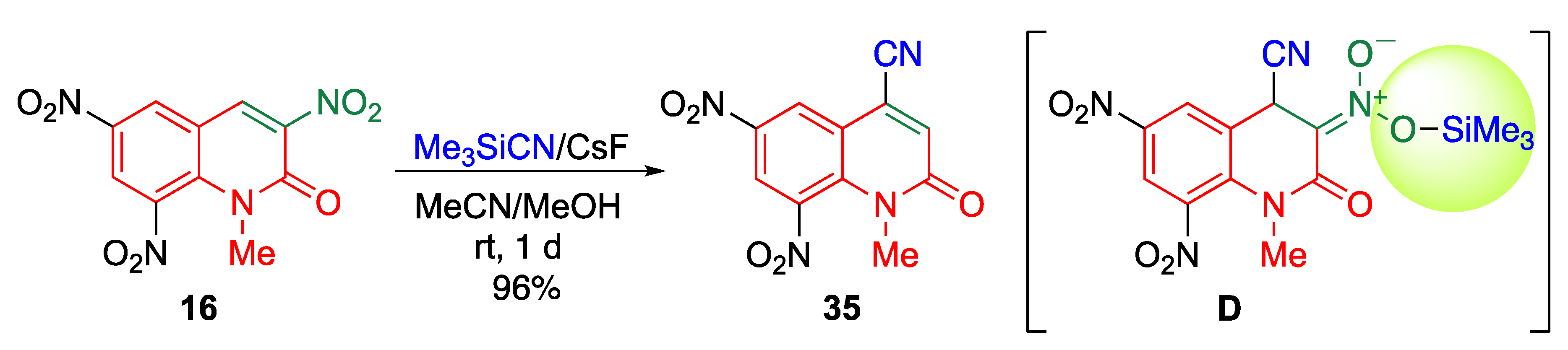
5.1.5. cine-Substitution of Trinitroquinolone with Cyanides
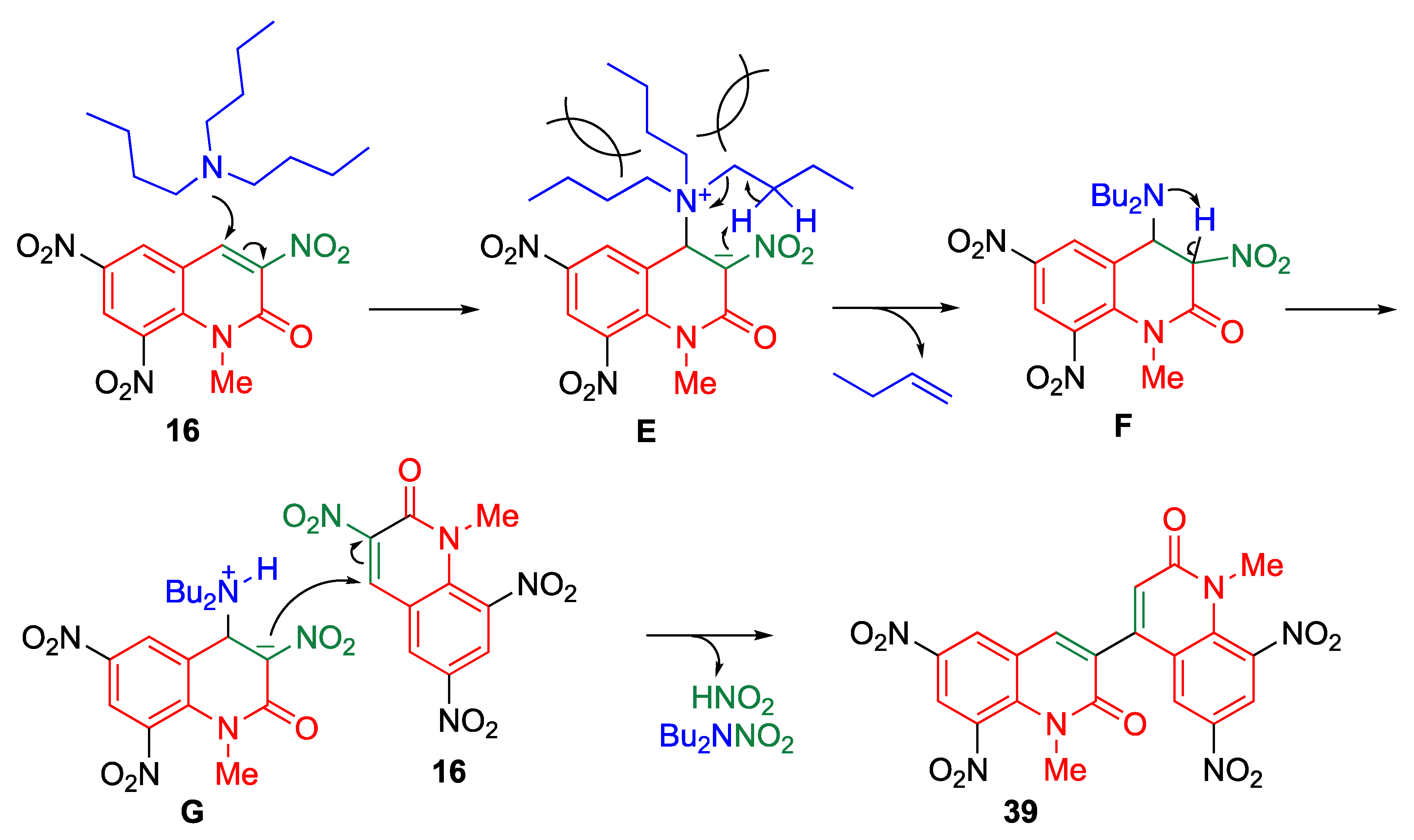
5.1.6. Reaction of Trinitroquinolone with Tertiary Amines
5.2. Direct C–N Bond Formation at the 4-Position
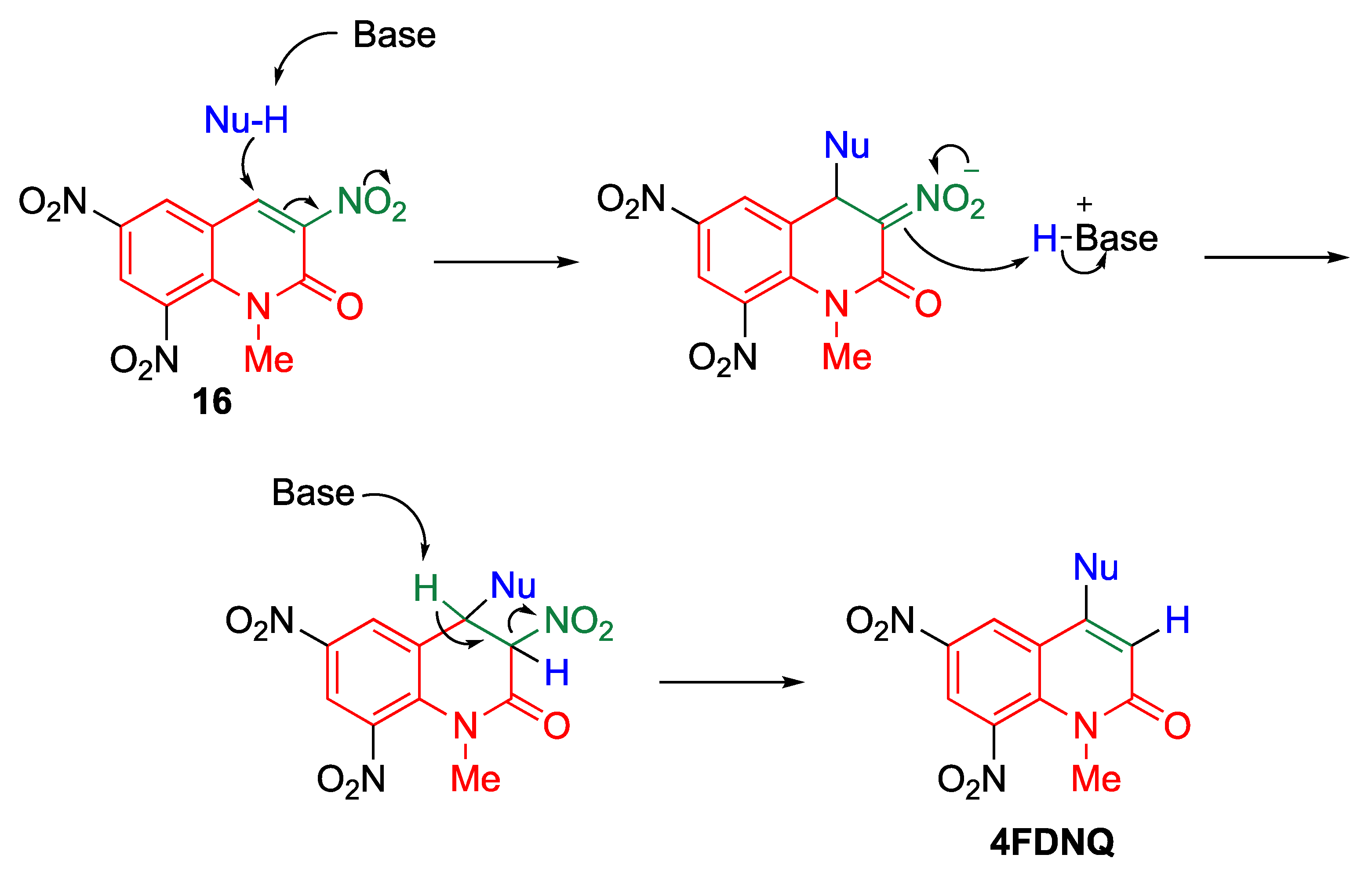
5.2.1. cine-Substitution of Trinitroquinolone with Primary Amines
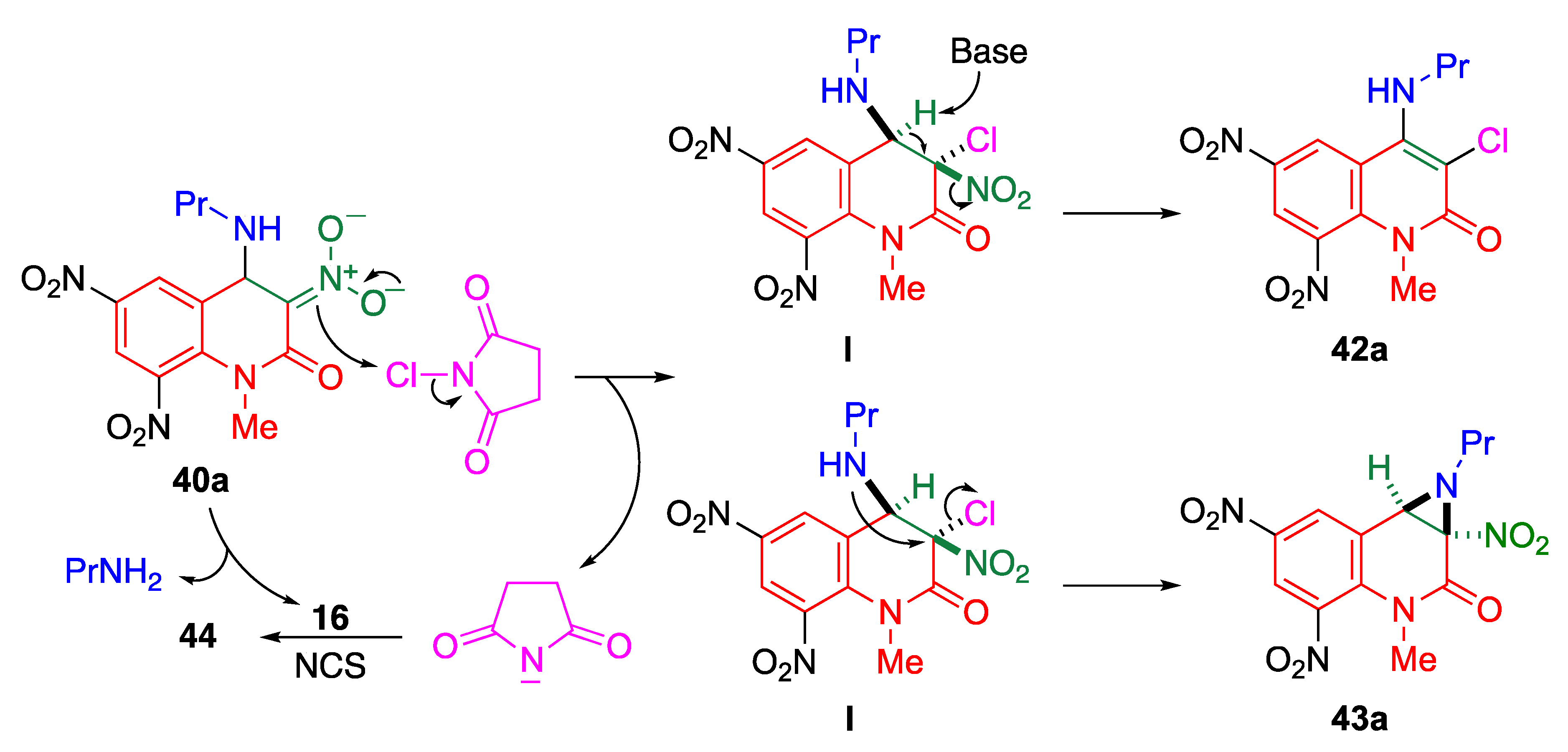
5.2.2. Amino-Halogenation and Imido-Halogenation of Quinolones
5.2.3. Aziridination of Quinolones
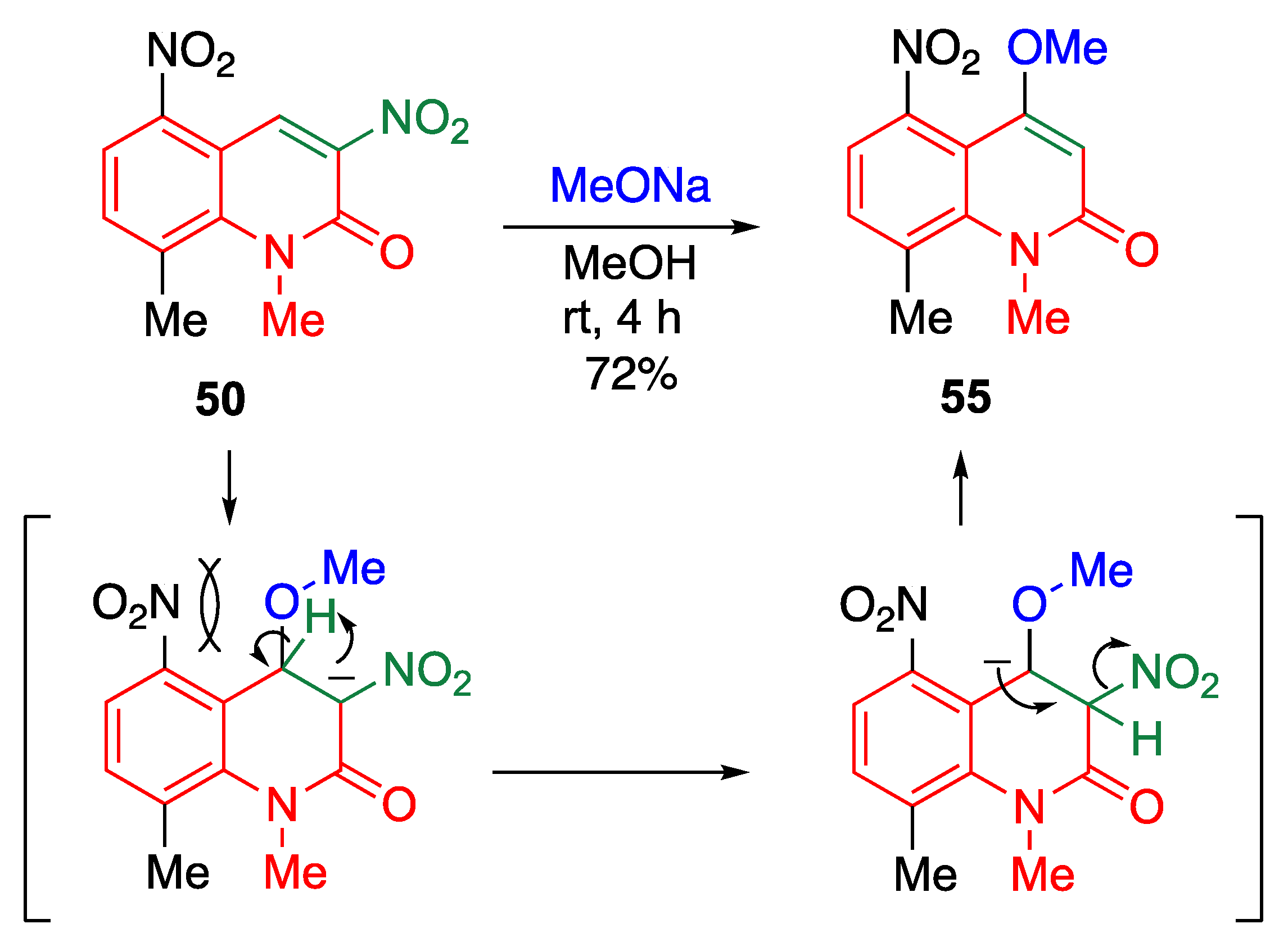
5.3. Direct C–O Bond Formation at the 4-Position
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Asif, M. A Mini Review: Biological Significances of Nitrogen Hetero Atom Containing Heterocyclic Compounds. Int. J. Bioorg. Chem. 2017, 2, 146–152. [Google Scholar]
- Joule, J.A. Chapter Four-Natural Products Containing Nitrogen Heterocycles-Some Highlights 1990–2015. Adv. Heterocycl. Chem. 2016, 119, 81–106. [Google Scholar]
- Kvasnica, M.; Urban, M.; Dickinson, N.J.; Sarek, J. Pentacyclic Triterpenoids with Nitrogen- and Sulfur-Containing Heterocycles: Synthesis and Medicinal Significance. Nat. Prod. Rep. 2015, 32, 1303–1330. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.M.; Sperry, J. Natural Products Containing a Nitrogen–Nitrogen Bond. J. Nat. Prod. 2013, 76, 794–812. [Google Scholar] [CrossRef] [PubMed]
- Zeidler, J.; Baraniak, D.; Ostrowski, T. Bioactive Nucleoside Analogues Possessing Selected Five-Membered Azaheterocyclic Bases. Eur. J. Med. Chem. 2015, 97, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Miura, M. A Lesson for Site-Selective C-H Functionalization on 2-Pyridones: Radical, Organometallic, Directing Group and Steric Controls. Chem. Sci. 2018, 9, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Akihisa, T.; Yokokawa, S.; Ogihara, E.; Matsumoto, M.; Zhang, J.; Kikuchi, T.; Koike, K.; Abe, M. Melanogenesis-Inhibitory and Cytotoxic Activities of Limonoids, Alkaloids, and Phenolic Compounds from Phellodendron amurense Bark. Chem. Biodiversity 2017, 14, e1700105. [Google Scholar] [CrossRef]
- Seya, K.; Miki, I.; Murata, K.; Junke, H.; Motomura, S.; Araki, T.; Itoyama, Y.; Oshima, Y. Pharmacological Properties of Pteleprenine, a Quinoline Alkaloid Extracted from Orixa Japonica, on Guinea-Pig Ileum and Canine Left Atrium. J. Pharm. Pharmacol. 1998, 507, 803–807. [Google Scholar] [CrossRef]
- Kamikawa, T.; Hanaoka, Y.; Fujie, S.; Saito, K.; Yamagiwa, Y.; Fukuhara, K.; Kubo, I. SRS-A Antagonist Pyranoquinolone Alkaloids from East African Fagara Plants and Their Synthesis. Bioorg. Med. Chem. 1996, 4, 1317–1320. [Google Scholar] [CrossRef]
- Joseph, B.; Darro, F.; Béhard, A.; Lesur, B.; Collignon, F.; Decaestecker, C.; Frydman, A.; Guillaumet, G.; Kiss, R. 3-Aryl-2-Quinolone Derivatives: Synthesis and Characterization of In Vitro and In Vivo Antitumor Effects with Emphasis on a New Therapeutical Target Connected with Cell Migration. J. Med. Chem. 2002, 45, 2543–2555. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, F.; Jia, A.; Li, X. Palladium-Catalyzed Selective Oxidative Olefination and Arylation of 2-Pyridones. Chem. Sci. 2012, 3, 3231–3236. [Google Scholar] [CrossRef]
- Nakatani, A.; Hirano, K.; Satoh, T.; Miura, M. Nickel-Catalyzed Direct Alkylation of Heterocycles with α-Bromo Carbonyl Compounds: C3-Selective Functionalization of 2-Pyridones. Chem. Eur. J. 2013, 19, 7691–7695. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Min, Q.; Zhao, H.; Gu, J.; Zhang, X. A General Synthesis of Fluoroalkylated Alkenes by Palladium-Catalyzed Heck-Type Reaction of Fluoroalkyl Bromides. Angew. Chem. Int. Ed. 2014, 53, 1–6. [Google Scholar]
- Nakatani, A.; Hirano, K.; Satoh, T.; Miura, M. Manganese-Mediated C3-Selective Direct Alkylation and Arylation of 2-Pyridones with Diethyl Malonates and Arylboronic Acids. J. Org. Chem. 2014, 79, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Elenicha, O.V.; Lytvyn, R.Z.; Skripskaya, O.V.; Lyavinets, O.S.; Pitkovych, K.E.; Yagodinets, P.I.; Obushak, M.D. Synthesis of Nitrogen-Containing Heterocycles on the Basis of 3-(4-Acetylphenyl)-1-methylquinolin-2(1H)-one. Russ. J. Org. Chem. 2016, 52, 373–378. [Google Scholar] [CrossRef]
- Li, J.; Hu, D.; Liang, X.; Wang, Y.; Wang, H.; Pan, Y. Praseodymium(III)-Catalyzed Regioselective Synthesis of C3-N-Substituted Coumarins with Coumarins and Azides. J. Org. Chem. 2017, 82, 9006–9011. [Google Scholar] [CrossRef]
- Prendergast, A.M.; McGlacken, G.P. Transition Metal Mediated C–H Activation of 2-Pyrones, 2-Pyridones, 2-Coumarins and 2-Quinolones. Eur. J. Org. Chem. 2018, 6068–6082. [Google Scholar] [CrossRef]
- Maity, S.; Das, D.; Sarkar, S.; Samanta, R. Direct Pd(II)-Catalyzed Site-Selective C5-Arylation of 2-Pyridone Using Aryl Iodides. Org. Lett. 2018, 20, 5167–5171. [Google Scholar] [CrossRef]
- Diesel, J.; Finogenova, A.M.; Cramer, N. Nickel-Catalyzed Enantioselective Pyridone C–H Functionalizations Enabled by a Bulky N-Heterocyclic Carbene Ligand. J. Am. Chem. Soc. 2018, 140, 4489–4493. [Google Scholar] [CrossRef]
- Hazra, S.; Hirano, K.; Miura, M. Solvent-Controlled Rhodium-Catalyzed C6-Selective C–H Alkenylation and Alkynylation of 2-Pyridones with Acrylates. Asian J. Org. Chem. 2019, 8, 1097–1101. [Google Scholar] [CrossRef]
- Zhao, H.; Xu, X.; Luo, Z.; Cao, L.; Li, B.; Li, H.; Xu, L.; Fan, Q.; Walsh, P.J. Rhodium(I)-Catalyzed C6-Selective C–H Alkenylation and Polyenylation of 2-Pyridones with Alkenyl and Conjugated Polyenyl Carboxylic Acids. Chem. Sci. 2019, 10, 10089–10096. [Google Scholar] [CrossRef]
- Nishiwaki, N. Chemistry of Nitroquinolones and Synthetic Application to Unnatural 1-Methyl-2-quinolone Derivatives. Molecules 2010, 15, 5174–5195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderari, G.; Seebach, D. Asymmetric Michael-Additions. Stereoselective Alkylation of Chiral, Non-racemic Enolates by Nitroolefins. Preparation of Enantiomerically Pure γ-Aminobutyric and Succinic Acid Derivatives. Helv. Chim. Acta 1985, 68, 1592–1604. [Google Scholar] [CrossRef]
- Ballini, R.; Bosica, G.; Fiorini, D.; Petrini, M. Unprecedented, Selective Nef Reaction of Secondary Nitroalkanes Promoted by DBU under Basic Homogeneous Conditions. Tetrahedron Lett. 2002, 43, 5233–5235. [Google Scholar] [CrossRef]
- Wehrli, P.A.; Schaer, B. Direct Transformation of Primary Nitro Compounds into Nitriles. New Syntheses of α,β-Unsaturated Nitriles and Cyanohydrin Acetates. J. Org. Chem. 1977, 42, 3956–3958. [Google Scholar] [CrossRef]
- Orlandi, M.; Brenna, D.; Harms, R.; Jost, S.; Benaglia, M. Recent Developments in the Reduction of Aromatic and Aliphatic Nitro Compounds to Amines. Org. Process Res. Dev. 2018, 22, 430–445. [Google Scholar] [CrossRef]
- Yadav, M.R.; Nagaoka, M.; Kashihara, M.; Zhong, R.-L.; Miyazaki, T.; Sakai, S.; Nakao, Y. The Suzuki–Miyaura Coupling of Nitroarenes. J. Am. Chem. Soc. 2017, 139, 9423–9426. [Google Scholar] [CrossRef]
- Inoue, F.; Kashihara, M.; Yadav, M.R.; Nakao, Y. Buchwald–Hartwig Amination of Nitroarenes. Angew. Chem. Int. Ed. 2017, 56, 13307–13309. [Google Scholar] [CrossRef]
- Halimehjani, A.Z.; Namboothiri, I.N.N.; Hooshmand, S.E. Nitroalkenes in the Synthesis of Carbocyclic Compounds. RSC Adv. 2014, 4, 31261–31299. [Google Scholar] [CrossRef]
- Hao, F.; Nishiwaki, N. Chemistry of Nitroaziridines. Heterocycles 2019, 99, 54–72. [Google Scholar]
- Hao, F.; Yokoyama, S.; Nishiwaki, N. Direct Dihalo-Alkoxylation of Nitroalkenes Leading to β,β-Dihalo-β-nitroethyl Alkyl Ethers. Org. Biomol. Chem. 2018, 16, 2768–2775. [Google Scholar] [CrossRef] [PubMed]
- Asahara, H.; Sofue, A.; Kuroda, Y.; Nishiwaki, N. Alkynylation and Cyanation of Alkenes Using Diverse Properties of a Nitro Group. J. Org. Chem. 2018, 83, 13691–13699. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, E.J.; Bakke, J.M.; Sletvold, I.; Svensen, H. Nucleophilic Alkylations of 3-Nitropyridines. Org. Biomol. Chem. 2004, 2, 2671–2676. [Google Scholar] [CrossRef] [PubMed]
- Fujita, R.; Watanabe, K.; Nishiuchi, Y.; Honda, R.; Matsuzaki, H.; Hongo, H. Diels-Alder Reactions of Nitro-2(1H)-pyridones with 2,3-Dimethyl-1,3-butadiene. Chem. Pharm. Bull. 2001, 49, 601–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, R.; Yoshisuji, T.; Wakayanagi, S.; Wakamatsu, H.; Matsuzaki, H. Synthesis of 5(6H)-Phenanthridones Using Diels-Alder Reaction of 3-Nitro-2(1H)-quinolones Acting Dienophiles. Chem. Pharm. Bull. 2006, 54, 204–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, R.; Watanabe, K.; Yoshisuji, T.; Kabuto, C.; Matsuzaki, H.; Hongo, H. Diels-Alder Reaction of 2(1 H)-quinolones Having an Electron-Withdrawing Group at the 3-Position Acting as Dienophiles with Dienes. Chem. Pharm. Bull. 2001, 49, 893–899. [Google Scholar] [CrossRef] [Green Version]
- Asahara, M.; Nagamatsu, M.; Tohda, Y.; Nishiwaki, N.; Ariga, M. Diels-Alder Reaction of 1-Methyl-3,6,8-trinitro-2-quinolone. J. Heterocycl. Chem. 2004, 41, 803–805. [Google Scholar] [CrossRef]
- Asahara, M.; Shibano, C.; Koyama, K.; Tamura, M.; Tohda, Y.; Ariga, M. The Nitroalkene Showing Dual Behaviors in the Same Reaction System. Tetrahedron Lett. 2005, 46, 7519–7521. [Google Scholar] [CrossRef]
- Nishiwaki, N.; Tanaka, C.; Asahara, M.; Asaka, N.; Tohda, Y.; Ariga, M. A Nitro Group Distorting 2-Quinolone Skeleton. Heterocycles 1999, 51, 567–574. [Google Scholar]
- Murashima, T.; Nishi, K.; Nakamoto, K.; Kato, A.; Tamai, R.; Uno, H.; Ono, N. Preparation of Novel Heteroisoindoles from Nitropyridines and Nitropyridones. Heterocycles 2002, 58, 301–310. [Google Scholar] [CrossRef]
- Nishiwaki, N.; Tanaka, A.; Uchida, M.; Tohda, Y.; Ariga, M. cine-Substitution of 1-Methyl-3,6,8-trinitro-2-quinolone. Bull. Chem. Soc. Jpn. 1996, 69, 1337–1381. [Google Scholar] [CrossRef]
- Chen, X.; Kobiro, K.; Asahara, H.; Kakiuchi, K.; Sugimoto, R.; Saigo, K.; Nishiwaki, N. Reactive 2-Quinolones Dearomatized by Steric Repulsion between 1-Methyl and 8-Substituted Groups. Tetrahedron 2013, 69, 4624–4630. [Google Scholar] [CrossRef] [Green Version]
- Asahara, M.; Ohtsutsumi, M.; Ariga, M.; Nishiwaki, N. Regioselective Nitroalkylation of the 1-Methyl-2-quinolone Framework. Heterocycles 2009, 78, 2851–2854. [Google Scholar]
- Asahara, M.; Katayama, T.; Tohda, Y.; Nishiwaki, N.; Ariga, M. Synthesis of Unnatural 1-Methyl-2-quinolone Derivatives. Chem. Pharm. Bull. 2004, 52, 1134–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasahara, M.; Ohtsutsumi, M.; Tamura, M.; Nishiwaki, N.; Ariga, M. Electrophilic Arylation of Phenols: Construction of a New Family of 1-Methyl-2-quinolones. Bull. Chem. Soc. Jpn. 2005, 78, 2235–2237. [Google Scholar] [CrossRef]
- Fleming, F.F.; Yao, L.; Ravikumar, P.C.; Funk, L.; Shook, B.C. Nitrile-Containing Pharmaceuticals: Efficacious Roles of the Nitrile Pharmacophore. J. Med. Chem. 2010, 53, 7902–7917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Li, Y.; Xiang, S.; Fan, W.; Jin, J.; Huang, D. Utilization of Nitriles as the Nitrogen Source: Practical and Economical Construction of 4-Aminopyrimidine and β-Enaminonitrile Skeletons. Org. Chem. Front. 2019, 6, 3071–3077. [Google Scholar] [CrossRef]
- Ghosh, T.; Si, A.; Misra, A.K. Facile Transformation of Nitriles into Thioamides: Application to C-Glycosyl Nitrile Derivatives. ChemistrySelect 2017, 2, 1366–1369. [Google Scholar] [CrossRef]
- Xi, F.; Kamal, F.; Schenerman, M.A. A Novel and Convenient Transformation of Nitriles to Aldehydes. Tetrahedron Lett. 2002, 43, 1395–1396. [Google Scholar] [CrossRef]
- Nishiwaki, N.; Sakashita, M.; Azuma, M.; Tanaka, C.; Tamura, M.; Asaka, N.; Hori, K.; Tohda, Y.; Ariga, M. Novel Functionalization of 1-Methyl-2-quinolone; Dimerization and Denitration of Trinitroquinolone. Tetrahedron 2002, 58, 473–478. [Google Scholar] [CrossRef]
- Asahara, M.; Nagamatsu, M.; Tohda, Y.; Nishiwaki, N.; Ariga, M. Effective C-N Bond Formation on the 1-Methyl-2-quinolone Skeleton. ARKIVOC 2005, 1, 1–6. [Google Scholar]
- Hao, F.; Asahara, H.; Nishiwaki, N. Direct Amino-halogenation and Aziridination of the 2-Quinolone Framework by Sequential Treatment of 3-Nitro-2-quinolone with Amine and N-Halosuccinimide. Tetrahedron 2017, 73, 1255–1264. [Google Scholar] [CrossRef]
- Hao, F.; Asahara, H.; Nishiwaki, N. A Direct and Vicinal Functionalization of the 1-Methyl-2-quinolone Framework: 4-Alkoxylation and 3-Chlorination. Org. Biomol. Chem. 2016, 14, 5128–5135. [Google Scholar] [CrossRef] [PubMed]




























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| R1 | R3 | Yield/% | |
|---|---|---|---|
| H | H | a | 26 |
| Me | H | b | 30 |
| Me | COOMe | c | 22 |

| R3 | Yield/% | ||
|---|---|---|---|
| 5 | 6 | ||
| H | a | 27 | 10 |
| NO2 | b | 33 | 15 |

| R1 | R4 | Yield/% | ||
|---|---|---|---|---|
| 8 | 9 | |||
| H | H | a | 0 | 15 |
| Me | H | b | 20 | 22 |
| Me | COOMe | c | 36 | 0 |

| R1 | R5 | Yield/% | |||
|---|---|---|---|---|---|
| 11 | 12 | 13 | |||
| H | NO2 | a | 13 | 15 | 0 |
| Me | NO2 | b1 | 36 | 33 | 0 |
| Me | COOMe | c | 31 | 0 | 14 |
| H | COOMe | d | 13 | 0 | 5 |

| R | R1 | R2 | R3 | Yield/% | |
|---|---|---|---|---|---|
| H | OMe | H | H | a | 83 |
| NO2 | OMe | H | H | b | 68 |
| H | H | Me | Me | c | 95 |
| NO2 | H | Me | Me | d | 64 |
| H | H | OMe | OMe | e | 45 |
| NO2 | H | OMe | OMe | f | 13 |

| R1 | R2 | Yield/% | |
|---|---|---|---|
| Me | Me | a | 88 |
| -(CH2)3- | b | 68 | |
| Me | OEt | c | 93 |
| CH2COOEt | OEt | d | 26 |
| OEt | OEt | e | 93 |

| R | Yield/% |
|---|---|
| NO2 | 88 |
| H | 0 1 |
| Me | 92 |

| R1 | R2 | Yield/% |
|---|---|---|
| Me | H | 80 |
| Et | H | 98 |
| Me | Me | 77 1 |

| R1 | R2 | R3 | Yield/% |
|---|---|---|---|
| H | Me | Me | 41 |
| Me | H | H | 83 |
| Ph | H | H | 83 |
| Et | Me | H | 18 |
| -(CH2)4- | H | 82 | |
| Ph | Me | H | 77 |
| Ph | Ph | H | 69 |
| 2-furyl | H | H | 45 |
| 2-pyridyl | H | H | 74 |

| R1 | R2 | R3 | Yield/% |
|---|---|---|---|
| H | Me | Me | 98 |
| -(CH2)4- | H | 40 | |
| Ph | Me | H | 43 |
| Ph | Ph | H | 98 |

| R5 | R6 | R7 | Yield/% | |
|---|---|---|---|---|
| NO2 | H | NO2 | a | 83 |
| NO2 | H | H | b | 47 |
| H | NO2 | H | c | quant. |

| R1 | R2 | R3 | Yield/% |
|---|---|---|---|
| Me | Me | Me | 0 |
| Et | Et | Et | 34 |
| Pr | Pr | Pr | 76 |
| Bu | Bu | Bu | 93 |
| Bu | Bu | Me | 79 |
| Bu | Me | Me | 18 |
| PhCH2 | PhCH2 | PhCH2 | 0 |

| R | Yield/% | ||
|---|---|---|---|
| 40 | 41 | ||
| Pr | a | 71 | 36 |
| i-Bu | b | 74 | 29 |
| s-Bu | c | 56 | 0 |
| t-Bu | d | 74 | 0 |

| R1 | R2 | Yield/% | |
|---|---|---|---|
| Pr | H | a | 62 |
| i-Bu | H | c | 70 |
| s-Bu | H | d | 49 |
| PhCH2 | H | e | 54 |
| HOCH2CH2 | H | f | 56 |
| CH2=CHCH2 | H | g | 35 |
| Ph | H | h | 54 |
| 4-MeOC6H4 | H | i | 37 |
| 4-BuC6H4 | H | j | 41 |
| 4-IC6H4 | H | k | 62 |
| 4-NO2C6H4 | H | l | trace |
| Et | Et | m | 0 |
| -(CH2)2-O-(CH2)2- | n | 62 | |

| X | Yield/% | |
|---|---|---|
| 45 | 46 | |
| Cl | 62 | 0 |
| Br | 63 | 16 |
| I | 0 | 62 |

| R1 | R6 | R7 | R8 | Yield/% | ||
|---|---|---|---|---|---|---|
| 48 | 49 | |||||
| Me | NO2 | H | NO2 | a | 62 | 0 |
| Me | NO2 | H | Me | b | 13 | 21 |
| Me | NO2 | H | H | c | 13 | 49 |
| Me | Br | H | H | d | trace | 68 |
| Me | H | H | H | e | 0 | 65 |
| Me | H | H | H | f | 0 | 71 |
| H | H | H | H | g | 0 | 61 |

| R | R6 | R8 | Yield/% | ||
|---|---|---|---|---|---|
| 52 | 53 | ||||
| Me | NO2 | NO2 | a | 81 | 85 |
| Et | NO2 | NO2 | b | quant. | 73 |
| i-Bu | NO2 | NO2 | c | - | 46 |
| i-Pr | NO2 | NO2 | d | - | 45 |
| PhCH2CH2 | NO2 | NO2 | e | - | 55 |
| CH2=CHCH2 | NO2 | NO2 | f | - | 51 |
| HC≡CCH2 | NO2 | NO2 | g | - | 29 |
| Me | NO2 | Me | h | - | 78 |
| Me | NO2 | H | i | - | 73 |
| Me | H | H | j | - | 65 |

| X | Yield/% | |
|---|---|---|
| 53 | 54 | |
| Cl | 85 | 0 |
| Br | 62 | 27 |
| I | 0 | 29 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, F.; Nishiwaki, N. Recent Progress in Nitro-Promoted Direct Functionalization of Pyridones and Quinolones. Molecules 2020, 25, 673. https://doi.org/10.3390/molecules25030673
Hao F, Nishiwaki N. Recent Progress in Nitro-Promoted Direct Functionalization of Pyridones and Quinolones. Molecules. 2020; 25(3):673. https://doi.org/10.3390/molecules25030673
Chicago/Turabian StyleHao, Feiyue, and Nagatoshi Nishiwaki. 2020. "Recent Progress in Nitro-Promoted Direct Functionalization of Pyridones and Quinolones" Molecules 25, no. 3: 673. https://doi.org/10.3390/molecules25030673
APA StyleHao, F., & Nishiwaki, N. (2020). Recent Progress in Nitro-Promoted Direct Functionalization of Pyridones and Quinolones. Molecules, 25(3), 673. https://doi.org/10.3390/molecules25030673