
PNA Clamping in Nucleic Acid Amplification Protocols to Detect Single Nucleotide Mutations Related to Cancer

Abstract
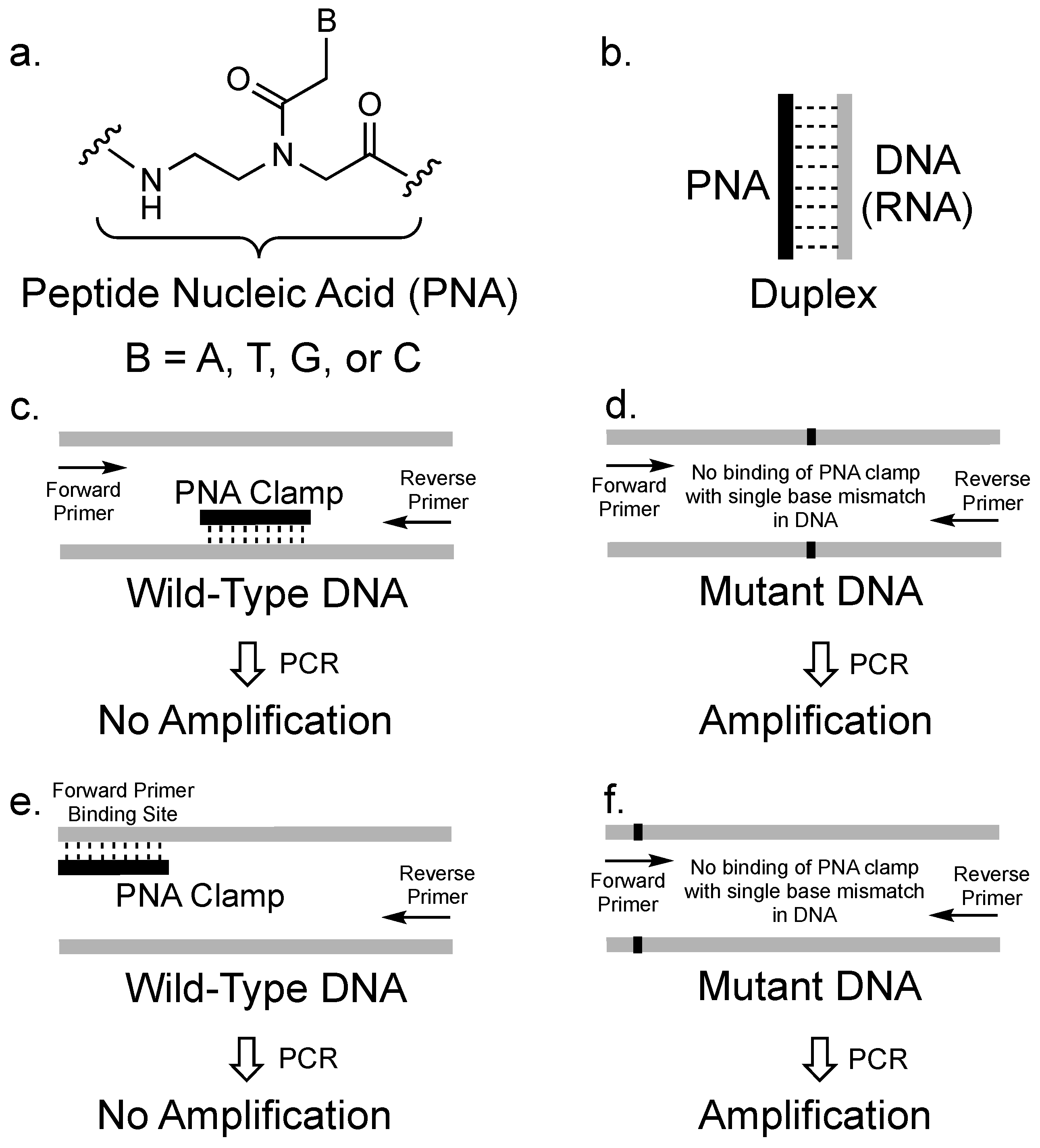
:1. Introduction
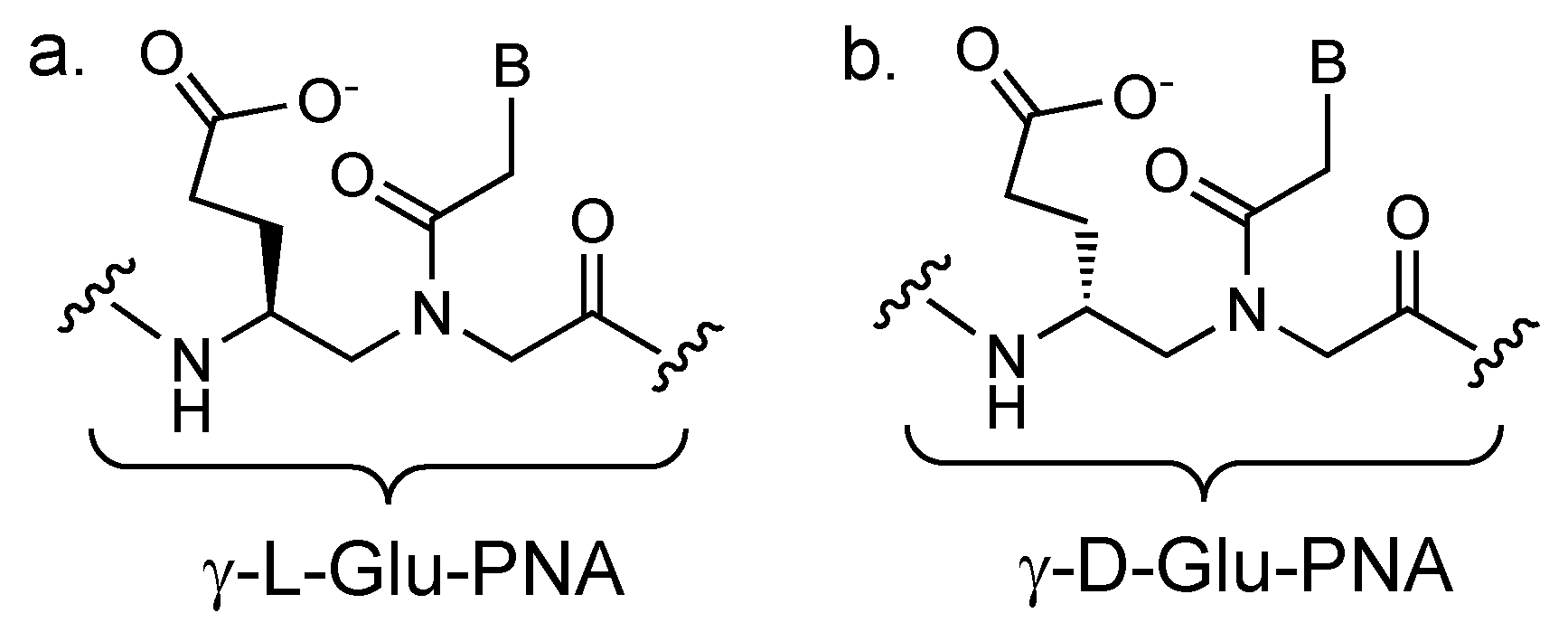
2. The Concept of PCR Clamping via PNA
3. PCR Clamping via PNA to Detect Mutated DNA in Cancer
4. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Syvänen, A.C. Accessing genetic variation: Genotyping single nucleotide polymorphisms. Nat. Rev. Genet. 2001, 2, 930–942. [Google Scholar] [CrossRef] [PubMed]
- Craig Venter, J.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Park, S.E.; Kim, M.; Ji, Y.I.; Kang, M.Y.; Jung, E.H.; Ko, E.; Kim, Y.; Kim, S.; Shim, Y.M.; et al. A functional single nucleotide polymorphism at the promoter region of cyclin A2 is associated with increased risk of colon, liver, and lung cancers. Cancer 2011, 117, 4080–4091. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.W.; Ye, Y.; Lin, J.; Vaporciyan, A.A.; Roth, J.A.; Wu, X. Genetic Variations in Epigenetic Genes Are Predictors of Recurrence in Stage I or II Non-Small Cell Lung Cancer Patients. Clin. Cancer Res. 2012, 18, 585–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mates, I.N.; Jinga, V.; Csiki, I.E.; Mates, D.; Dinu, D.; Constantin, A.; Jinga, M. Single nucleotide polymorphisms in colorectal cancer: associations with tumor site and TNM stage. J. Gastrointestin. Liver Dis. 2012, 21, 45–52. [Google Scholar] [PubMed]
- Park, C.; Han, S.; Lee, K.-M.; Choi, J.-Y.; Song, N.; Jeon, S.; Park, S.K.; Ahn, H.S.; Shin, H.Y.; Kang, H.J.; et al. Association between CASP7 and CASP14 genetic polymorphisms and the risk of childhood leukemia. Hum. Immunol. 2012, 73, 736–739. [Google Scholar] [CrossRef]
- Han, S.; Lan, Q.; Park, A.K.; Lee, K.-M.; Park, S.K.; Ahn, H.S.; Shin, H.Y.; Kang, H.J.; Koo, H.H.; Seo, J.J.; et al. Polymorphisms in innate immunity genes and risk of childhood leukemia. Hum. Immunol. 2010, 71, 727–730. [Google Scholar] [CrossRef] [Green Version]
- Penas-Steinhardt, A.; Tellechea1, M.L.; Gomez-Rosso, L.; Brites, F.; Frechtel, G.D.; Poskus, E. Association of common variants in JAK2 gene with reduced risk of metabolic syndrome and related disorders. BMC Med. Genet. 2011, 12, 166. [Google Scholar] [CrossRef] [Green Version]
- Oguro, R.; Kamide, K.; Katsuya, T.; Akasaka, H.; Sugimoto, K.; Congrains, A.; Arai, Y.; Hirose, N.; Saitoh, S.; Ohishi, M.; et al. A single nucleotide polymorphism of the adenosine deaminase, RNA-specific gene is associated with the serum triglyceride level, abdominal circumference, and serum adiponectin concentration. Exp. Gerontol. 2012, 47, 183–187. [Google Scholar] [CrossRef]
- Sanda, S.; Wei, S.; Rue, T.; Shilling, H.; Greenbaum, C. A SNP in G6PC2 predicts insulin secretion in type 1 diabetes. Acta Diabetol. 2013, 50, 459–462. [Google Scholar] [CrossRef]
- Stark, K.; Reinhard, W.; Grassl, M.; Erdmann, J.; Schunkert, H.; Illig, T.; Hengstenberg, C. Common Polymorphisms Influencing Serum Uric Acid Levels Contribute to Susceptibility to Gout, but Not to Coronary Artery Disease. PLoS ONE 2009, 4, e7729. [Google Scholar] [CrossRef] [PubMed]
- Aomori, T.; Yamamoto, K.; Oguchi-Katayama, A.; Kawai, Y.; Ishidao, T.; Mitani, Y.; Kogo, Y.; Lezhava, A.; Fujita, Y.; Obayashi, K.; et al. Rapid Single-Nucleotide Polymorphism Detection of Cytochrome P450 (CYP2C9) and Vitamin K Epoxide Reductase (VKORC1) Genes for the Warfarin Dose Adjustment by the SMart-Amplification Process Version 2. Clin. Chem. 2009, 55, 804–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, T.; Miyata, T. Warfarin dose and the pharmacogenomics of CYP2C9 and VKORC1 — Rationale and perspectives. Thromb. Res. 2007, 120, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Xie, L.; Song, X. The diagnostic accuracy of circulating free DNA for the detection of KRAS mutation status in colorectal cancer: A meta-analysis. Cancer Med. 2019, 8, 1218–1231. [Google Scholar] [CrossRef] [PubMed]
- Beganovic, S. Clinical Significance of the KRAS Mutation. Bosn. J. Basic Med. Sci. 2010, 9, S17–S20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgensztern, D.; Politi, K.; Herbst, R.S. EGFR Mutations in Non–Small-Cell Lung Cancer. JAMA Oncol. 2015, 1, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Cunha Santos, G.; Shepherd, F.A.; Tsao, M.S. EGFR Mutations and Lung Cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 49–69. [Google Scholar] [CrossRef] [Green Version]
- Petitjean, A.; Achatz, M.I.W.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [Green Version]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Huo, X.; Tang, C.; Ye, H.; Nandakumar, V.; Lou, F.; Zhang, D.; Jiang, S.; Sun, H.; Dong, H.; et al. Frequent KIT Mutations in Human Gastrointestinal Stromal Tumors. Sci. Rep. 2015, 4, 5907. [Google Scholar] [CrossRef] [Green Version]
- Bando, H.; Yoshino, T.; Tsuchihara, K.; Ogasawara, N.; Fuse, N.; Kojima, T.; Tahara, M.; Kojima, M.; Kaneko, K.; Doi, T.; et al. KRAS mutations detected by the amplification refractory mutation system–Scorpion assays strongly correlate with therapeutic effect of cetuximab. Br. J. Cancer 2011, 105, 403–406. [Google Scholar] [CrossRef]
- Kimura, T.; Okamoto, K.; Miyamoto, H.; Kimura, M.; Kitamura, S.; Takenaka, H.; Muguruma, N.; Okahisa, T.; Aoyagi, E.; Kajimoto, M.; et al. Clinical Benefit of High-Sensitivity KRAS Mutation Testing in Metastatic Colorectal Cancer Treated with Anti-EGFR Antibody Therapy. Oncology 2012, 82, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, F.; Blanchard, F.; Charbonnier, F.; Le Pessot, F.; Lamy, A.; Galais, M.P.; Bastit, L.; Killian, A.; Sesboüé, R.; Tuech, J.J.; et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br. J. Cancer 2007, 96, 1166–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tougeron, D.; Lecomte, T.; Pages, J.C.; Villalva, C.; Collin, C.; Ferru, A.; Tourani, J.M.; Silvain, C.; Levillain, P.; Karayan-Tapon, L. Effect of low-frequency KRAS mutations on the response to anti-EGFR therapy in metastatic colorectal cancer. Ann. Oncol. 2013, 24, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K. PCR-Based Detection Methods for Single-Nucleotide Polymorphism or Mutation. In Advances in Clinical Chemistry; Elsevier: New York, NY, USA, 2017; Volume 9, pp. 45–72. [Google Scholar]
- Harlé, A.; Busser, B.; Rouyer, M.; Harter, V.; Genin, P.; Leroux, A.; Merlin, J.-L. Comparison of COBAS 4800 KRAS, TaqMan PCR and High Resolution Melting PCR assays for the detection of KRAS somatic mutations in formalin-fixed paraffin embedded colorectal carcinomas. Virchows Arch. 2013, 462, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Brophy, V.H.; Cao, J.; Velez, M.; Hoeppner, C.; Soviero, S.; Lawrence, H.J. Analytical performance of a PCR assay for the detection of KRAS mutations (codons 12/13 and 61) in formalin-fixed paraffin-embedded tissue samples of colorectal carcinoma. Virchows Arch. 2012, 460, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsiatis, A.C.; Norris-Kirby, A.; Rich, R.G.; Hafez, M.J.; Gocke, C.D.; Eshleman, J.R.; Murphy, K.M. Comparison of Sanger Sequencing, Pyrosequencing, and Melting Curve Analysis for the Detection of KRAS Mutations. J. Mol. Diagn. 2010, 12, 425–432. [Google Scholar] [CrossRef]
- Fox, J.; England, J.; White, P.; Ellison, G.; Callaghan, K.; Charlesworth, N.; Hehir, J.; McCarthy, T.; Smith-Ravin, J.; Talbot, I.; et al. The detection of K-ras mutations in colorectal cancer using the amplification-refractory mutation system. Br. J. Cancer 1998, 77, 1267–1274. [Google Scholar] [CrossRef] [Green Version]
- Pinto, P.; Rocha, P.; Veiga, I.; Guedes, J.; Pinheiro, M.; Peixoto, A.; Pinto, C.; Fragoso, M.; Sanches, E.; Araújo, A.; et al. Comparison of methodologies for KRAS mutation detection in metastatic colorectal cancer. Cancer Genet. 2011, 204, 439–446. [Google Scholar] [CrossRef]
- Mitani, Y.; Lezhava, A.; Kawai, Y.; Kikuchi, T.; Oguchi-Katayama, A.; Kogo, Y.; Itoh, M.; Miyagi, T.; Takakura, H.; Hoshi, K.; et al. Rapid SNP diagnostics using asymmetric isothermal amplification and a new mismatch-suppression technology. Nat. Methods 2007, 4, 257–262. [Google Scholar] [CrossRef]
- Miyamae, Y.; Shimizu, K.; Mitani, Y.; Araki, T.; Kawai, Y.; Baba, M.; Kakegawa, S.; Sugano, M.; Kaira, K.; Lezhava, A.; et al. Mutation Detection of Epidermal Growth Factor Receptor and KRAS Genes Using the Smart Amplification Process Version 2 from Formalin-Fixed, Paraffin-Embedded Lung Cancer Tissue. J. Mol. Diagn. 2010, 12, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Taly, V.; Pekin, D.; Benhaim, L.; Kotsopoulos, S.K.; Le Corre, D.; Li, X.; Atochin, I.; Link, D.R.; Griffiths, A.D.; Pallier, K.; et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin. Chem. 2013, 59, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Dressman, D.; Yan, H.; Traverso, G.; Kinzler, K.W.; Vogelstein, B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl. Acad. Sci. USA 2003, 100, 8817–8822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thierry, A.R.; Mouliere, F.; El Messaoudi, S.; Mollevi, C.; Lopez-Crapez, E.; Rolet, F.; Gillet, B.; Gongora, C.; Dechelotte, P.; Robert, B.; et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat. Med. 2014, 20, 430–435. [Google Scholar] [CrossRef]
- Huang, J.F.; Zeng, D.Z.; Duan, G.J.; Shi, Y.; Deng, G.H.; Xia, H.; Xu, H.Q.; Zhao, N.; Fu, W.L.; Huang, Q. Single-tubed wild-type blocking quantitative PCR detection assay for the sensitive detection of codon 12 and 13 KRAS mutations. PLoS ONE 2015, 10, 1–23. [Google Scholar] [CrossRef]
- Mouliere, F.; El Messaoudi, S.; Pang, D.; Dritschilo, A.; Thierry, A.R. Multi-marker analysis of circulating cell-free DNA toward personalized medicine for colorectal cancer. Mol. Oncol. 2014, 8, 927–941. [Google Scholar] [CrossRef]
- Mouliere, F.; El Messaoudi, S.; Gongora, C.; Guedj, A.-S.; Robert, B.; Del Rio, M.; Molina, F.; Lamy, P.-J.; Lopez-Crapez, E.; Mathonnet, M.; et al. Circulating Cell-Free DNA from Colorectal Cancer Patients May Reveal High KRAS or BRAF Mutation Load. Transl. Oncol. 2013, 6, 319-IN8. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Diehl, F.; Dressman, D.; Vogelstein, B.; Kinzler, K.W. BEAMing up for detection and quantification of rare sequence variants. Nat. Methods 2006, 3, 95–97. [Google Scholar] [CrossRef]
- Baker, M. Digital PCR hits its stride. Nat. Methods 2012, 9, 541–544. [Google Scholar] [CrossRef]
- Didelot, A.; Kotsopoulos, S.K.; Lupo, A.; Pekin, D.; Li, X.; Atochin, I.; Srinivasan, P.; Zhong, Q.; Olson, J.; Link, D.R.; et al. Multiplex Picoliter-Droplet Digital PCR for Quantitative Assessment of DNA Integrity in Clinical Samples. Clin. Chem. 2013, 59, 815–823. [Google Scholar] [CrossRef] [Green Version]
- Murdock, D.G.; Wallace, D.C. PNA-Mediated PCR Clamping: Applications and Methods. In Peptide Nucleic Acids; Nielsen, P.E., Ed.; Humana Press: Totowa, NJ, USA, 2002; pp. 145–164. ISBN 978-1-59259-290-6. [Google Scholar]
- Nielsen, P.; Egholm, M.; Berg, R.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science. 1991, 254, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Nordén, B. Peptide nucleic acid (PNA): its medical and biotechnical applications and promise for the future. FASEB J. 2000, 14, 1041–1060. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E. Peptide Nucleic Acids; Humana Press: Totowa, NJ, USA, 2002; Volume 208, ISBN 1-59259-290-2. [Google Scholar]
- D’Agata, R.; Giuffrida, M.; Spoto, G. Peptide Nucleic Acid-Based Biosensors for Cancer Diagnosis. Molecules 2017, 22, 1951. [Google Scholar] [CrossRef] [Green Version]
- Shigi, N.; Sumaoka, J.; Komiyama, M. Applications of PNA-Based Artificial Restriction DNA Cutters. Molecules 2017, 22, 1586. [Google Scholar] [CrossRef] [Green Version]
- Appella, D.H. Overcoming biology’s limitations. Nat. Chem. Biol. 2010, 6, 87–88. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Appella, D.H. Advantages of Peptide Nucleic Acids as Diagnostic Platforms for Detection of Nucleic Acids in Resource-Limited Settings. J. Infect. Dis. 2010, 201, S42–S45. [Google Scholar] [CrossRef]
- Pellestor, F.; Paulasova, P. The peptide nucleic acids (PNAs), powerful tools for molecular genetics and cytogenetics. Eur. J. Hum. Genet. 2004, 12, 694–700. [Google Scholar] [CrossRef]
- Ørum, H.; Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O.; Stanley, C. Single base pair mutation analysis by PNA directed PCR clamping. Nucleic Acids Res. 1993, 21, 5332–5336. [Google Scholar] [CrossRef] [Green Version]
- Valones, M.A.A.; Guimarães, R.L.; Brandão, L.A.C.; de Souza, P.R.E.; de Carvalho, A.T.; Crovela, S. Principles and applications of polymerase chain reaction in medical diagnostic fields: a review. Braz. J. Microbiol. 2009, 40, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Jang, M.; Kim, J.; Park, H. Highly sensitive PNA Array Platform Technology for Single Nucleotide Mismatch Discrimination. J. Microbiol. Biotechnol. 2010, 20, 287–293. [Google Scholar] [CrossRef]
- Orum, H. PCR clamping. Curr. Issues Mol. Biol. 2000, 2, 27–30. [Google Scholar]
- Bishop, J.M. Molecular themes in oncogenesis. Cell 1991, 64, 235–248. [Google Scholar] [CrossRef]
- Lee, M.S.; Kopetz, S. Current and Future Approaches to Target the Epidermal Growth Factor Receptor and Its Downstream Signaling in Metastatic Colorectal Cancer. Clin. Colorectal Cancer 2015, 14, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Peeters, M.; Siena, S.; Humblet, Y.; Hendlisz, A.; Neyns, B.; Canon, J.L.; Van Laethem, J.L.; Maurel, J.; Richardson, G.; et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy- refractory metastatic colorectal cancer. J. Clin. Oncol. 2007, 25, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras Mutations and Benefit from Cetuximab in Advanced Colorectal Cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [Green Version]
- Thiede, C.; Bayerdorffer, E.; Blasczyk, R.; Wittig, B.; Neubauer, A. Simple and Sensitive Detection of Mutations in the Ras Proto-Oncogenes Using PNA-Mediated PCR Clamping. Nucleic Acids Res. 1996, 24, 983–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-Y.; Shiesh, S.-C.; Wu, S.-J. Rapid Detection of K-ras Mutations in Bile by Peptide Nucleic Acid-mediated PCR Clamping and Melting Curve Analysis: Comparison with Restriction Fragment Length Polymorphism Analysis. Clin. Chem. 2004, 50, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Taback, B.; Bilchik, A.J.; Saha, S.; Nakayama, T.; Wiese, D.A.; Turner, R.R.; Kuo, C.T.; Hoon, D.S.B. Peptide nucleic acid clamp PCR: A novel K-ras mutation detection assay for colorectal cancer micrometastases in lymph nodes. Int. J. Cancer 2004, 111, 409–414. [Google Scholar] [CrossRef]
- Däbritz, J.; Hänfler, J.; Preston, R.; Stieler, J.; Oettle, H. Detection of Ki-ras mutations in tissue and plasma samples of patients with pancreatic cancer using PNA-mediated PCR clamping and hybridisation probes. Br. J. Cancer 2005, 92, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.-D. Detection of rare mutant K-ras DNA in a single-tube reaction using peptide nucleic acid as both PCR clamp and sensor probe. Nucleic Acids Res. 2006, 34, e12. [Google Scholar] [CrossRef] [Green Version]
- Gilje, B.; Heikkilä, R.; Oltedal, S.; Tjensvoll, K.; Nordgård, O. High-Fidelity DNA Polymerase Enhances the Sensitivity of a Peptide Nucleic Acid Clamp PCR Assay for K-ras Mutations. J. Mol. Diagn. 2008, 10, 325–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, T.; Shimizu, K.; Nakamura, K.; Nakamura, T.; Mitani, Y.; Obayashi, K.; Fujita, Y.; Kakegawa, S.; Miyamae, Y.; Kaira, K.; et al. Usefulness of peptide nucleic acid (PNA)-clamp smart amplification process version 2 (SmartAmp2) for clinical diagnosis of KRAS codon12 mutations in lung adenocarcinoma: Comparison of PNA-clamp SmartAmp2 and PCR-related methods. J. Mol. Diagn. 2010, 12, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.E.; Lim, H.S.; An, C.H.; Jeong, E.G.; Han, J.Y.; Lee, S.H.; Yoo, N.J. Detection of Low-Level KRAS Mutations Using PNA-Mediated Asymmetric PCR Clamping and Melting Curve Analysis with Unlabeled Probes. J. Mol. Diagn. 2010, 12, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-T.; Kim, J.W.; Kim, S.K.; Joe, G.H.; Hong, I.S. Simultaneous Genotyping of Multiple Somatic Mutations by Using a Clamping PNA and PNA Detection Probes. ChemBioChem 2015, 16, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Englund, E.A.; Appella, D.H. γ-Substituted Peptide Nucleic Acids Constructed from L-Lysine are a Versatile Scaffold for Multifunctional Display. Angew. Chemie Int. Ed. 2007, 46, 1414–1418. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Rakhit, C.; Ottolini, B.; Jones, C.; Pringle, J.; Shaw, J.; Martins, L.M. Peptide nucleic acid clamping to improve the sensitivity of Ion Torrent-based detection of an oncogenic mutation in KRAS. Matters 2017, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Behn, M.; Schuermann, M. Sensitive detection of p53 gene mutations by a “mutant enriched” PCR-SSCP technique. Nucleic Acids Res. 1998, 26, 1356–1358. [Google Scholar] [CrossRef] [Green Version]
- Behn, M.; Thiede, C.; Neubauer, A.; Pankow, W.; Schuermann, M. Facilitated detection of oncogene mutations from exfoliated tissue material by a PNA-mediated enriched PCR protocol. J. Pathol. 2000, 190, 69–75. [Google Scholar] [CrossRef]
- Myal, Y.; Blanchard, A.; Watson, P.; Corrin, M.; Shiu, R.; Iwasiow, B. Detection of Genetic Point Mutations by Peptide Nucleic Acid-Mediated Polymerase Chain Reaction Clamping Using Paraffin-Embedded Specimens. Anal. Biochem. 2000, 285, 169–172. [Google Scholar] [CrossRef]
- Raben, D.; Helfrich, B.A.; Chan, D.; Johnson, G.; Bunn, P.A. ZD1839, a selective epidermal growth factor receptor tyrosine kinase inhibitor, alone and in combination with radiation and chemotherapy as a new therapeutic strategy in non–small cell lung cancer. Semin. Oncol. 2002, 29, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Miyazawa, H.; Huqun; Tanaka, T.; Udagawa, K.; Kato, M.; Fukuyama, S.; Yokote,, A.; Kobayashi, K.; Kanazawa, M.; et al. Genetic heterogeneity of the epidermal growth factor receptor in non-small cell lung cancer cell lines revealed by a rapid and sensitive detection system, the peptide nucleic acid-locked nucleic acid PCR clamp. Cancer Res. 2005, 65, 7276–7282. [Google Scholar]
- McTigue, P.M.; Peterson, R.J.; Kahn, J.D. Sequence-Dependent Thermodynamic Parameters for Locked Nucleic Acid (LNA)-DNA Duplex Formation. Biochemistry 2004, 43, 5388–5405. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, K.Y.; Kim, Y.-C.; Kim, K.-S.; Lee, S.Y.; Jang, T.W.; Lee, M.K.; Shin, K.-C.; Lee, G.H.; Lee, J.C.; et al. Detection and comparison of peptide nucleic acid-mediated real-time polymerase chain reaction clamping and direct gene sequencing for epidermal growth factor receptor mutations in patients with non-small cell lung cancer. Lung Cancer 2012, 75, 321–325. [Google Scholar] [CrossRef]
- Yam, I.; Lam, D.C.L.; Chan, K.; Chung-Man Ho, J.; Ip, M.; Lam, W.K.; Chan, T.K.; Chan, V. EGFR array: Uses in the detection of plasma EGFR mutations in non-small cell lung cancer patients. J. Thorac. Oncol. 2012, 7, 1131–1140. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-R.; Lee, S.; Hyun, D.-S.; Lee, M.; Lee, H.-K.; Choi, C.-M.; Yang, S.-H.; Kim, Y.-C.; Lee, Y.; Kim, S.; et al. Detection of EGFR mutations in circulating free DNA by PNA-mediated PCR clamping. J. Exp. Clin. Cancer Res. 2013, 32, 50. [Google Scholar] [CrossRef] [Green Version]
- Han, J.-Y.; Choi, J.-J.; Kim, J.Y.; Han, Y.L.; Lee, G.K. PNA clamping-assisted fluorescence melting curve analysis for detecting EGFR and KRAS mutations in the circulating tumor DNA of patients with advanced non-small cell lung cancer. BMC Cancer 2016, 16, 627. [Google Scholar]



{kind=link}
{kind=link}
| Oncogene | Method Used in Combination with PNA Clamped PCR | Mutated DNA Detected in Presence of Wild-Type DNA | PNA Sequence Length- Number of Nucleobases | Refs |
|---|---|---|---|---|
| KRAS | DNA Sequencing | 0.5% | 15 | [59] |
| Fluorescent Probes | 0.03% | 17 | [60] | |
| Melting Curve Analysis | 0.05%, 0.001% | 15,17 | [61,62] | |
| Fluorescent PNA Sensor with LightCycler | 0.01% | 17 | [63] | |
| High Fidelity DNA Polymerase | 0.005% | 17 | [64] | |
| SmartAmp2 | 1% | 17 | [65] | |
| Asymmetric PCR | 0.1% | 17 | [66] | |
| Modified PNA Detection Probes | 1% | 17 | [67] | |
| Next Generation Sequencing | N/A | 6 | [70] | |
| p53 | PCR-SSCP | N/A,0.1%,0.05% | 15,15,15 | [71,72,73] |
| EGFR | PNA+LNA | 0.1% | 14-18 | [75] |
| DNA Sequencing | 0.1% | Kit | [77] | |
| Fluorescent Melting Curve Analysis | 0.1%,N/A,0.01% | Kit | [78,79,80] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fouz, M.F.; Appella, D.H. PNA Clamping in Nucleic Acid Amplification Protocols to Detect Single Nucleotide Mutations Related to Cancer. Molecules 2020, 25, 786. https://doi.org/10.3390/molecules25040786
Fouz MF, Appella DH. PNA Clamping in Nucleic Acid Amplification Protocols to Detect Single Nucleotide Mutations Related to Cancer. Molecules. 2020; 25(4):786. https://doi.org/10.3390/molecules25040786
Chicago/Turabian StyleFouz, Munira F., and Daniel H. Appella. 2020. "PNA Clamping in Nucleic Acid Amplification Protocols to Detect Single Nucleotide Mutations Related to Cancer" Molecules 25, no. 4: 786. https://doi.org/10.3390/molecules25040786
APA StyleFouz, M. F., & Appella, D. H. (2020). PNA Clamping in Nucleic Acid Amplification Protocols to Detect Single Nucleotide Mutations Related to Cancer. Molecules, 25(4), 786. https://doi.org/10.3390/molecules25040786