Synthesis of Natural (−)-Antrocin and Its Enantiomer via Stereoselective Aldol Reaction



Abstract
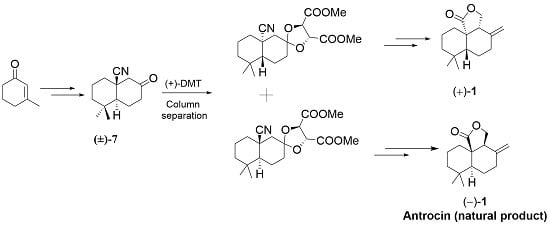
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
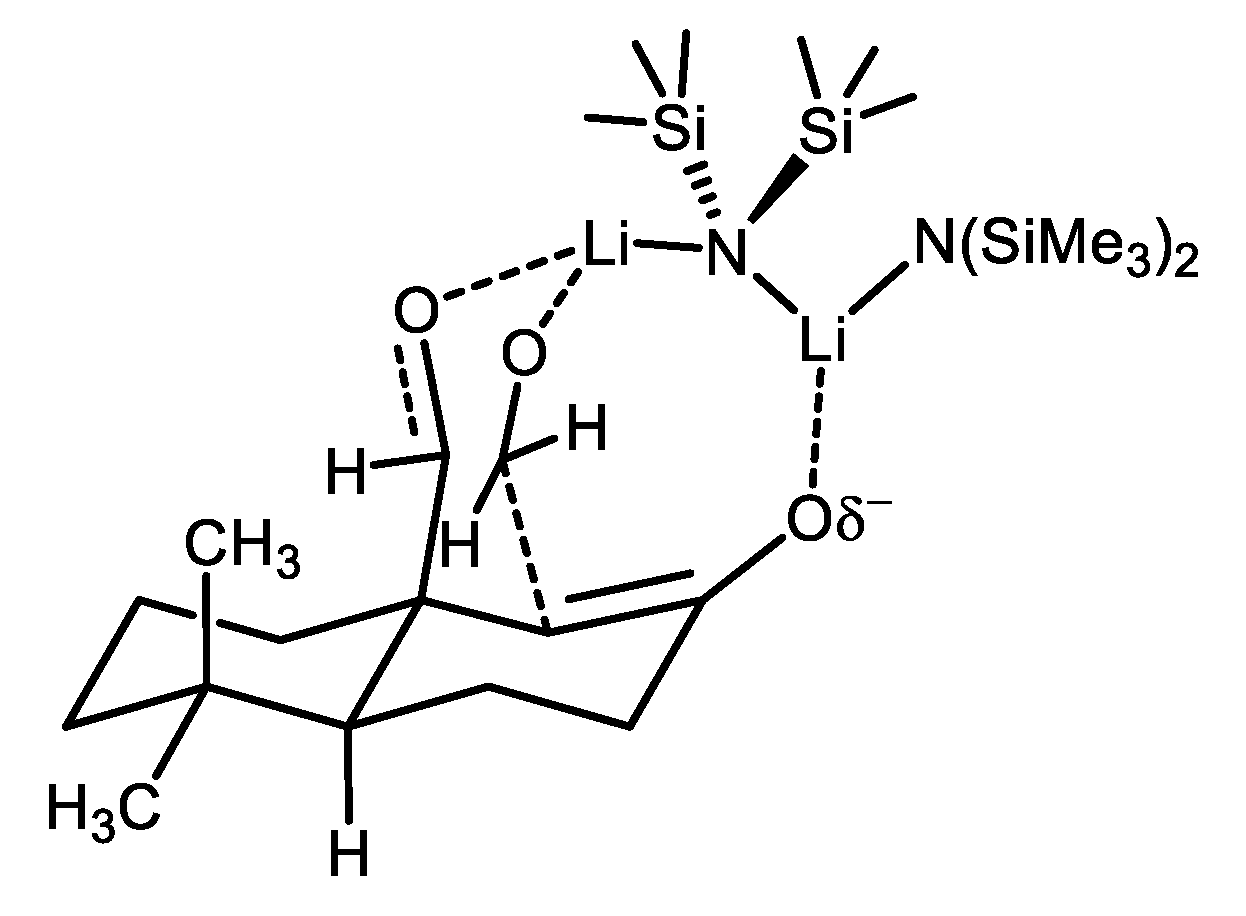{kind=link}
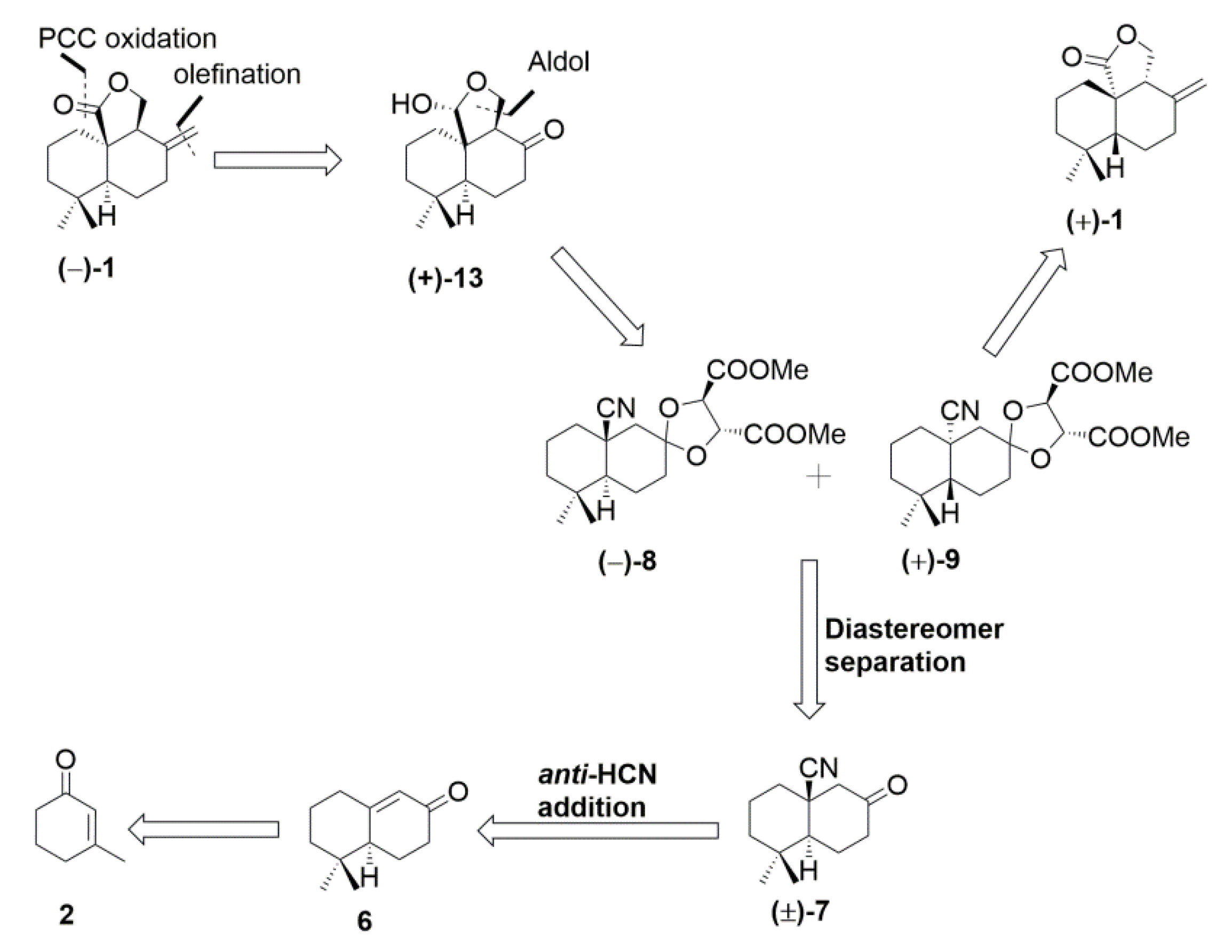{kind=link}
{kind=link}
{kind=link}
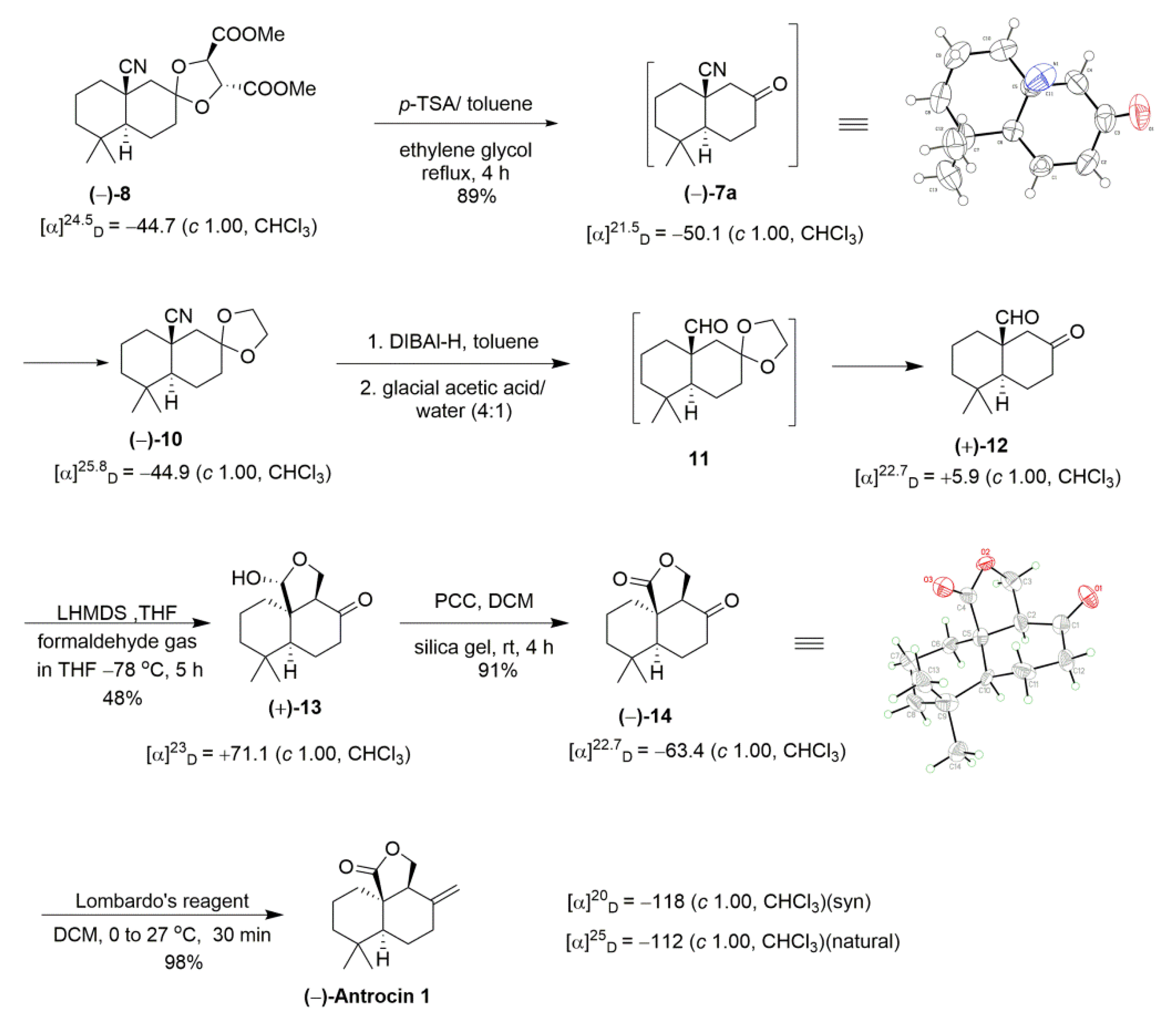{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
3. Conclusions
4. Experimental
General Conditions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Neerman, M.F. Sesquiterpene lactones: A diverse class of compounds found in essential oils possessing antibacterial and antifungal properties. Int. J. Aromatherapy 2003, 13, 114–120. [Google Scholar] [CrossRef]
- Seaman, F.C.; Funk, V.A. Cladistic analysis of complex natural products: Developing transformation series from sesquiterpene lactone data. Taxon 1983, 32, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Jansen, B.J.M.; de Groot, A. The occurrence and biological activity of drimane sesquiterpenoids. Nat. Prod. Rep. 1991, 8, 309–318. [Google Scholar] [CrossRef]
- Zhao, S.S.; Leung, K.S.-Y. Quality evaluation of mycelial Antrodia camphorata using high-performance liquid chromatography (HPLC) coupled with diode array detector and mass spectrometry (DAD-MS). Chin. Med. 2010, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-C.; Liu, Y.-W.; Ker, Y.-B.; Wu, Y.-Y.; Lai, E.Y.; Chyau, C.-C.; Hseu, T.-H.; Peng, R.Y. Chemical Characterization and Anti-inflammatory Effect of Polysaccharides Fractionated from Submerge-Cultured Antrodia camphorata Mycelia. J. Agric. Food Chem. 2007, 55, 5007–5012. [Google Scholar] [CrossRef] [PubMed]
- Geethangili, M.; Tzeng, Y.-M. Review of Pharmacological Effects of Antrodia camphorata and Its Bioactive Compounds. Evid Based Complement Alternat. Med. 2011, 2011, 212641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.C. King of Ganoderma: Antrodia camphorata in Taiwan, 2nd ed.; Yuen Chi Jai Book Publishing: Taipei, Taiwan, 2008. [Google Scholar]
- Wu, D.-P.; Chiang, H.-C. Constituents of Antrodia Cinnamomea. J. Chin. Chem. Soc. 1995, 42, 797–800. [Google Scholar] [CrossRef]
- Lee, I.-H.; Huang, R.-L.; Chen, C.-T.; Chen, H.-C.; Hsu, W.-C.; Lu, M.-K. Antrodia camphorata polysaccharides exhibit anti-hepatitis B virus effects. Microbiol. Lett. 2002, 209, 63–67. [Google Scholar] [CrossRef] [Green Version]
- Hseu, Y.-C.; Chen, S.-C.; Chen, H.-C.; Liao, J.-W.; Yang, H.-L. Antrodia camphorata inhibits proliferation of human breast cancer cells in vitro and in vivo. Food Chem. Toxicol. 2008, 46, 2680–2688. [Google Scholar] [CrossRef]
- Lee, Y.-P.; Tsai, W.-C.; Ko, C.-J.; Rao, Y.K.; Yang, C.-R.; Chen, D.-R.; Yang, M.-H.; Yang, C.-C.; Tzeng, Y.-M. Anticancer Effects of Eleven Triterpenoids Derived from Antrodia camphorata. Anticancer Res. 2012, 32, 2727–2734. [Google Scholar]
- Ao, Z.-H.; Xu, Z.-H.; Lu, Z.-M.; Xu, H.-Y.; Zhang, X.-M.; Dou, W.-F. Niuchangchih (Antrodia camphorata) and its potential in treating liver diseases. J. Ethnopharmacol. 2009, 121, 194–212. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-H.; Wu, A.T.H.; Tzeng, D.T.W.; Huang, C.-C.; Tzeng, Y.-M.; Chao, T.-Y. Antrocin, a bioactive component from Antrodia cinnamomea, suppresses breast carcinogenesis and stemness via downregulation of β-catenin/Notch1/Akt signaling. Phytomedicine 2019, 52, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Hung-Chen, C.; De-Peng, W.; Cherng, I.W.; Chuen-Her, U. A sesquiterpene lactone, phenyl and biphenyl compounds from Antrodia cinnamomea. Phytochemistry 1995, 39, 613–616. [Google Scholar] [CrossRef]
- Rao, Y.K.; Wu, A.T.H.; Geethangili, M.; Huang, M.-T.; Chao, W.-J.; Wu, C.-H.; Deng, W.-P.; Yeh, C.-T.; Tzeng, Y.-M. Identification of Antrocin from Antrodia camphorata as a Selective and Novel Class of Small Molecule Inhibitor of Akt/mTOR Signaling in Metastatic Breast Cancer MDA-MB-231 Cells. Chem. Res. Toxicol. 2011, 24, 238–245. [Google Scholar] [CrossRef]
- Chen, C.-C.; Shiao, Y.-J.; Lin, R.-D.; Shao, Y.-Y.; Lai, M.-N.; Lin, C.-C.; Ng, L.-T.; Kuo, Y.-H. Neuroprotective Diterpenes from the Fruiting Body of Antrodia camphorata. J. Nat. Prod. 2006, 69, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; Yang, H.O.; Kang, Y.-H.; Suh, D.-Y.; Go, H.J.; Song, W.-J.; Kim, Y.C.; Park, M.K. In Vitro Platelet-Activating Factor Receptor Binding Inhibitory Activity of Pinusolide Derivatives: A Structure−Activity Study. J. Med. Chem. 1998, 41, 2626–2630. [Google Scholar] [CrossRef]
- Chiang, Y.-M.; Liu, H.-K.; Lo, J.-M.; Chien, S.-C.; Chan, Y.-F.; Lee, T.-H.; Su, J.-K.; Kuo, Y.-H. Cytotoxic Constituents of the Leaves of Calocedrus Formosana. J. Chin. Chem. Soc. 2003, 50, 161–166. [Google Scholar] [CrossRef]
- Smith, L.; Watson, M.B.; O’Kane, S.L.; Drew, P.J.; Lind, M.J.; Cawkwell, L. The analysis of doxorubicin resistance in human breast cancer cells using antibody microarrays. Mol. Cancer. Ther. 2006, 5, 2115–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Fang, L.; Tan, C.; Shi, L.; Zhang, W.; Li, C.-C.; Luo, T.; Yang, Z. Total Syntheses of Drimane-Type Sesquiterpenoids Enabled by a Gold-Catalyzed Tandem Reaction. J. Am. Chem. Soc. 2011, 133, 14944–14947. [Google Scholar] [CrossRef]
- Li, F.-Z.; Li, S.; Zhang, P.-P.; Huang, Z.-H.; Zhang, W.-B.; Gong, J.; Yang, Z. A chiral pool approach for asymmetric syntheses of (−)-antrocin, (+)-asperolide C, and (−)-trans-ozic acid. Chem. Comm. 2016, 52, 12426–12429. [Google Scholar] [CrossRef]
- Huang, S.-H.; Liang, K.-H.; Lu, J.-S.; Chang, W.-S.; Su, M.-D.; Yang, T.-F. Total Synthesis of (+)-Antrocin and Its Diastereomer and Clarification of the Absolute Stereochemistry of (−)-Antrocin. J. Org. Chem. 2017, 82, 9576–9584. [Google Scholar] [CrossRef] [PubMed]
- Tai, D.-F.; Angamuthu, V.; Tzeng, Y.-M. Process for preparing optically active antrocin. U.S. Patent US0096324 A1, 18 April 2013. [Google Scholar]
- Hosmer, C.A.; Comber, R.N.; Brouillette, W.J. Syntheses of alpha.-, beta.-, and gamma.-substituted carnitines via beta.-keto esters. J. Org. Chem. 1985, 50, 3627–3631. [Google Scholar] [CrossRef]
- Feringa, B.L. Trimethylsilyl-Halide-Mediated Conjugated Addition to Allylic Acetals. Syn. Comm. 1985, 15, 87–89. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.M.; Nieto-Oberhuber, C.; Shair, M.D. Enantioselective Synthesis of (+)-Cortistatin A, a Potent and Selective Inhibitor of Endothelial Cell Proliferation. J. Am. Chem. Soc. 2008, 130, 16864–16866. [Google Scholar] [CrossRef] [PubMed]
- House, H.O.; Wilkins, J.M. Reactions involving electron transfer. 12. Effects of solvent and substituents upon the ability of lithium diorganocuprates to add to enones. J. Org. Chem. 1978, 43, 2443–2454. [Google Scholar] [CrossRef]
- Spessard, S.J.; Stoltz, B.M. Progress toward the Synthesis of Garsubellin A and Related Phloroglucins: The Direct Diastereoselective Synthesis of the Bicyclo[3.3.1]nonane Core. Org. Lett. 2002, 4, 1943–1946. [Google Scholar] [CrossRef] [Green Version]
- Taber, D.F.; Sikkander, M.I.; Storck, P.H. Enantioselective Synthesis of (+)-Majusculone. J. Org. Chem. 2007, 72, 4098–4101. [Google Scholar] [CrossRef] [Green Version]
- Nagata, W.; Yoshioka, M.; Hirai, S. Hydrocyanation. IV. New hydrocyanation methods using hydrogen cyanide and an alkylaluminum, and an alkylaluminum cyanide. J. Am. Chem. Soc. 1972, 94, 4635–4643. [Google Scholar] [CrossRef]
- Ihara, M.; Katsumata, A.; Egashira, M.; Suzuki, S.; Tokunaga, Y.; Fukumoto, K. Stereoselective Construction of the Diterpene Part of Indole Alkaloids, Radarins, by Way of Intramolecular Diels-Alder Reaction. J. Org. Chem. 1995, 60, 5560–5566. [Google Scholar] [CrossRef]
- Suryawanshi, S.N.; Fuchs, P.L. Bruceantin support studies. 10. Use of an axial alpha.-face control element in intramolecular conjugate additions: Synthesis of an ABCD tetracyclic bruceantin precursor. J. Org. Chem. 1986, 51, 902–921. [Google Scholar] [CrossRef]
- Utimoto, K.; Wakabayashi, Y.; Horiie, T.; Inoue, M.; Shishiyama, Y.; Obayashi, M.; Nozaki, H. Cyanotrimethylsilane as a versatile reagent for introducing cyanide functionality. Tetrahedron 1983, 39, 967–973. [Google Scholar] [CrossRef]
- Dailey, O.D.; Fuchs, P.L. Synthesis of a model for the BCE ring system of bruceantin. A caveat on the cyclohexene fwdarw. trans diaxial diol conversion. J. Org. Chem. 1980, 45, 216–236. [Google Scholar] [CrossRef]
- Furuichi, N.; Hata, T.; Soetjipto, H.; Kato, M.; Katsumura, S. Common synthetic strategy for optically active cyclic terpenoids having a 1,1,5-trimethyl-trans-decalin nucleus: Syntheses of (+)-acuminolide, (−)-spongianolide A, and (+)-scalarenedial. Tetrahedron 2001, 57, 8425–8442. [Google Scholar] [CrossRef]
- Cha, J.S.; Kim, E.J.; Kwon, O.O.; Kim, J.M. Chemoselective Reduction of Carbonyl Compounds with Diisopinocampheylchloroborane. Synlett 1995, 1995, 331–332. [Google Scholar] [CrossRef]
- Hori, K.; Hikage, N.; Inagaki, A.; Mori, S.; Nomura, K.; Yoshii, E. Total synthesis of tetronomycin. J. Org. Chem. 1992, 57, 2888–2902. [Google Scholar] [CrossRef]
- Zoretic, P.A.; Zhang, Y.; Fang, H.; Ribeiro, A.A.; Dubay, G. Advanced Tetracycles in a Stereoselective Approach to d,l-Spongiatriol and Related Metabolites: The Use of Radicals in the Synthesis of Angular Electrophores. J. Org. Chem. 1998, 63, 1162–1167. [Google Scholar] [CrossRef]
- Schlosser, M.; Jenny, T.; Guggisberg, Y. Monomeric Formaldehyde in Ethereal Solution. Synlett 1990, 1990, 704. [Google Scholar] [CrossRef]
- Kato, M.; Matsumura, Y.; Heima, K.; Fukamiya, N.; Kabuto, C.; Yoshikoshi, A. Total synthesis of dl-siccanin and dl-siccanochromene E. Tetrahedron 1987, 43, 711–722. [Google Scholar] [CrossRef]
- NOE spectroscopic date given in Supplementary Materials.
- Díaz, S.; Cuesta, J.; González, A.; Bonjoch, J. Synthesis of (−)-Nakamurol A and Assignment of Absolute Configuration of Diterpenoid (+)-Nakamurol, A. J. Org. Chem. 2003, 68, 7400–7406. [Google Scholar] [CrossRef]
- Toshima, H.; Oikawa, H.; Toyomasu, T.; Sassa, T. Total Synthesis of (+)-Albicanol and (+)-Albicanyl Acetate. Biosci. Biotechnol. Biochem. 2001, 65, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Skepper, C.K.; Quach, T.; Molinski, T.F. Total Synthesis of Enigmazole A from Cinachyrella enigmatica. Bidirectional Bond Constructions with an Ambident 2,4-Disubstituted Oxazole Synthon. J. Am. Chem. Soc. 2010, 132, 10286–10292. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Electronic Supplementary Information (ESI) available: Spectral data (1H, 13C NMR, and HRMS) of all compounds; CIF for (−)-7, (−)-8, and (−)-14 compounds crystallographic data material are available from the authors. |












© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angamuthu, V.; Tai, D.-F. Synthesis of Natural (−)-Antrocin and Its Enantiomer via Stereoselective Aldol Reaction. Molecules 2020, 25, 831. https://doi.org/10.3390/molecules25040831
Angamuthu V, Tai D-F. Synthesis of Natural (−)-Antrocin and Its Enantiomer via Stereoselective Aldol Reaction. Molecules. 2020; 25(4):831. https://doi.org/10.3390/molecules25040831
Chicago/Turabian StyleAngamuthu, Venkatachalam, and Dar-Fu Tai. 2020. "Synthesis of Natural (−)-Antrocin and Its Enantiomer via Stereoselective Aldol Reaction" Molecules 25, no. 4: 831. https://doi.org/10.3390/molecules25040831
APA StyleAngamuthu, V., & Tai, D. -F. (2020). Synthesis of Natural (−)-Antrocin and Its Enantiomer via Stereoselective Aldol Reaction. Molecules, 25(4), 831. https://doi.org/10.3390/molecules25040831