Understanding the Pyrimethamine Drug Resistance Mechanism via Combined Molecular Dynamics and Dynamic Residue Network Analysis



Abstract
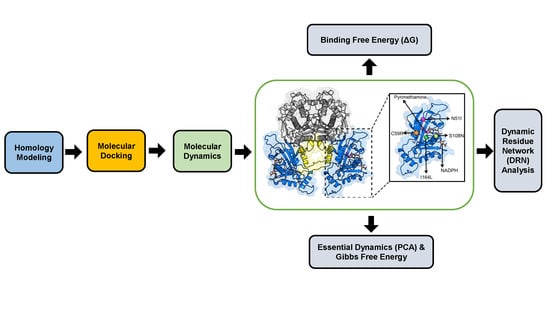
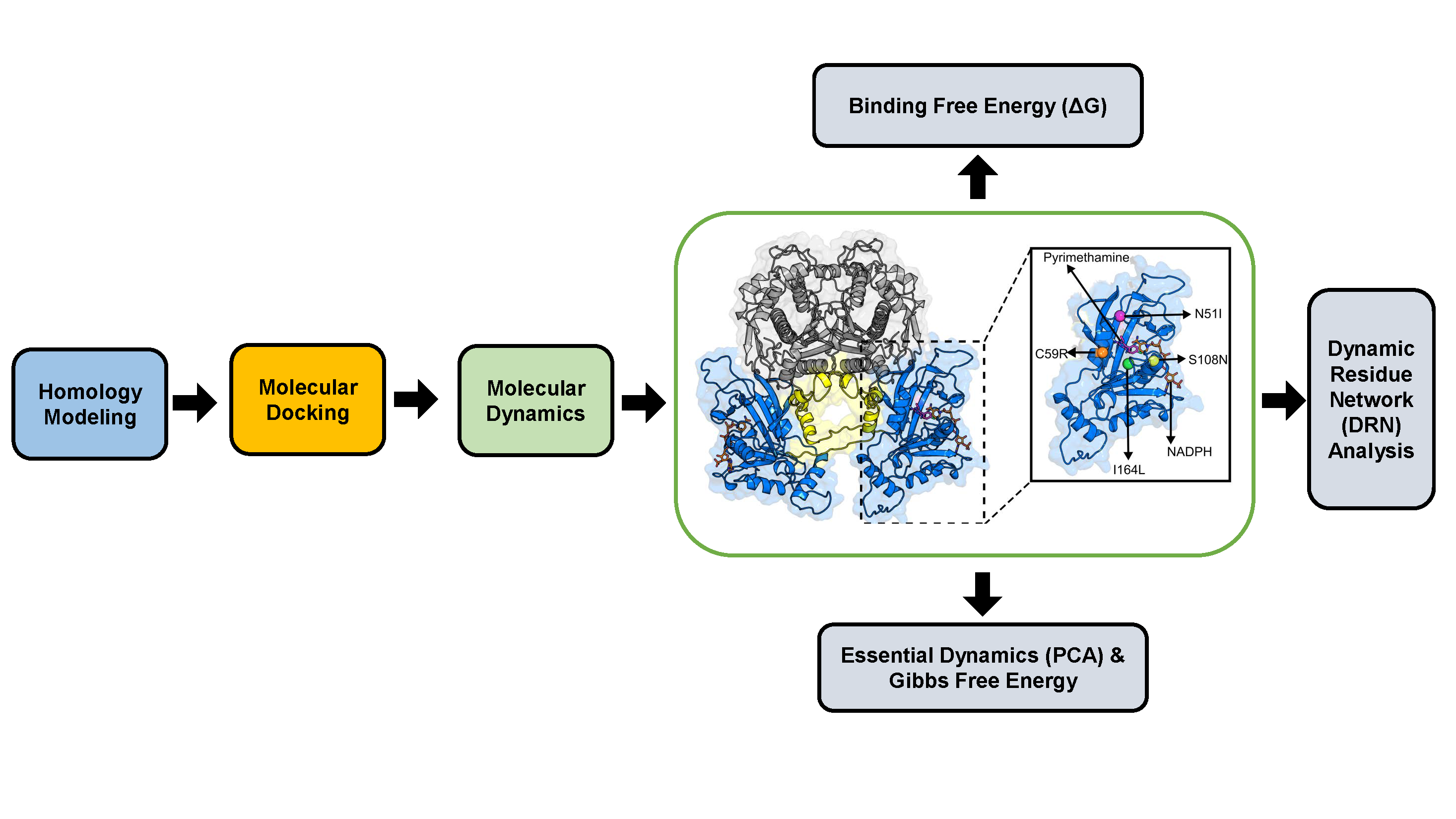
:
1. Introduction
2. Results and Discussion
2.1. Pyrimethamine Docked Differently to Protein with Resistance Mutations due to the Changes in the Active Site
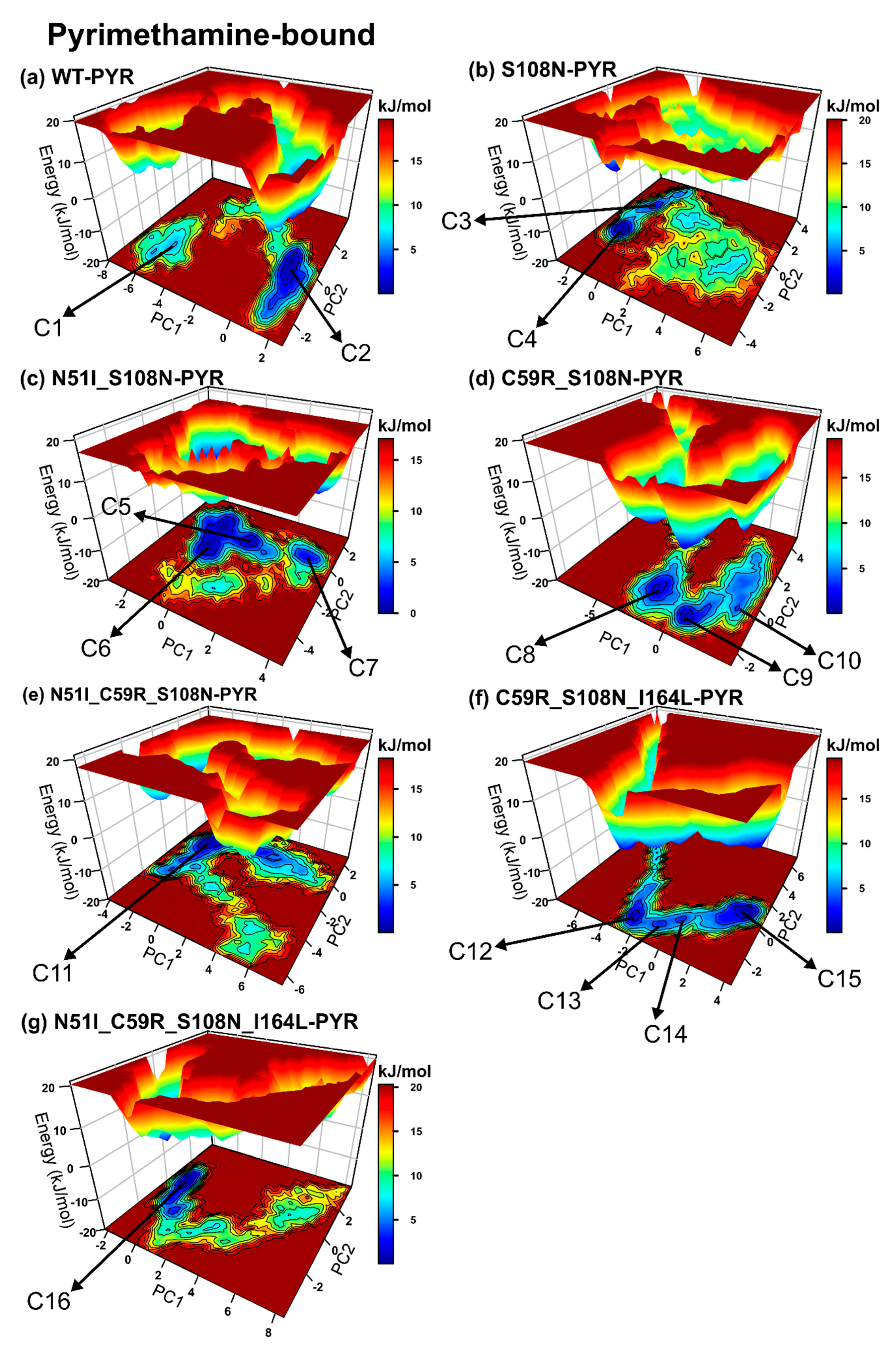
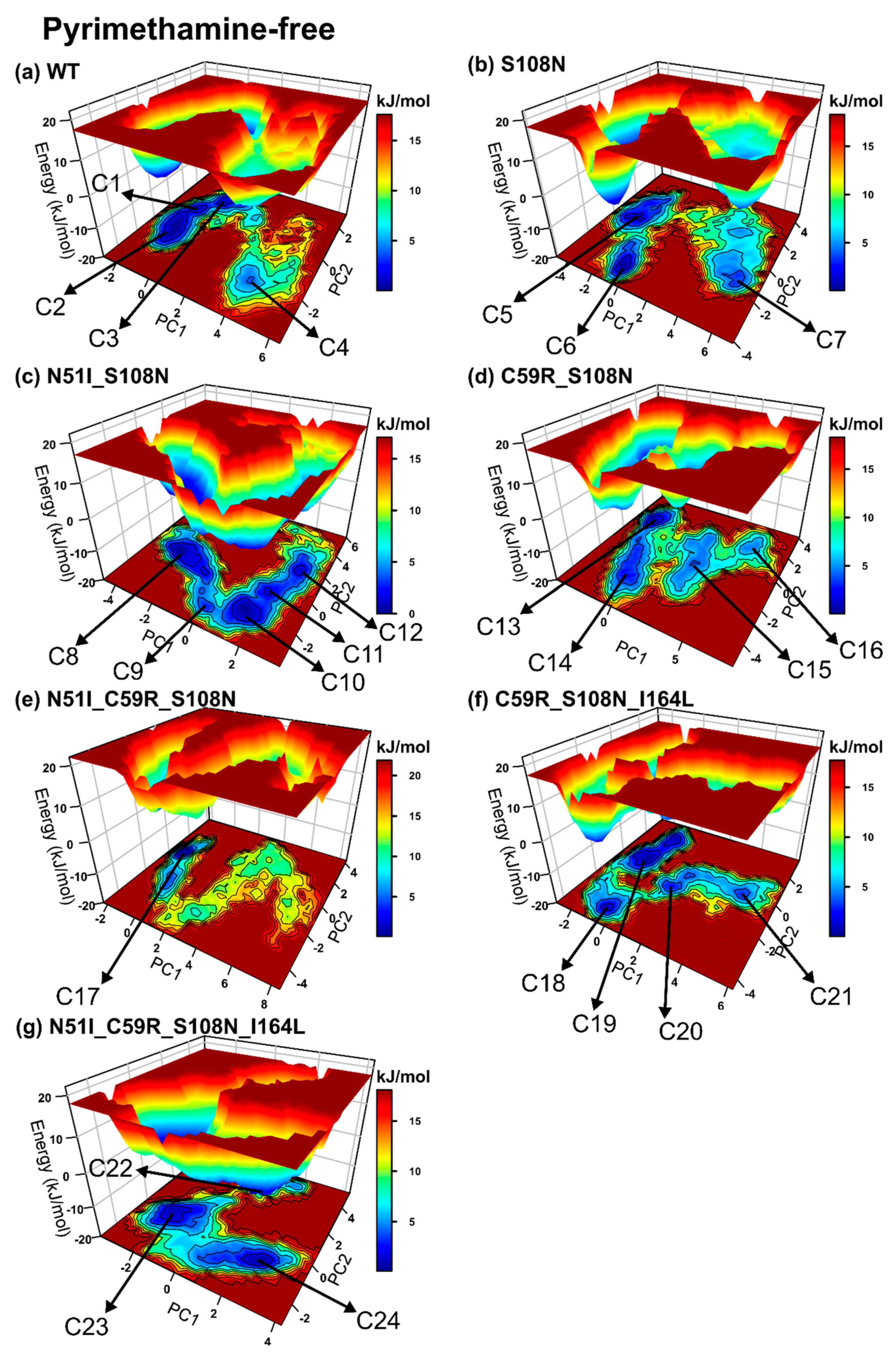
2.2. Global Analysis Revealed Differences in the Conformational Spaces Between WT and Proteins with Resistance Mutations in the Absence and Presence of the Drug
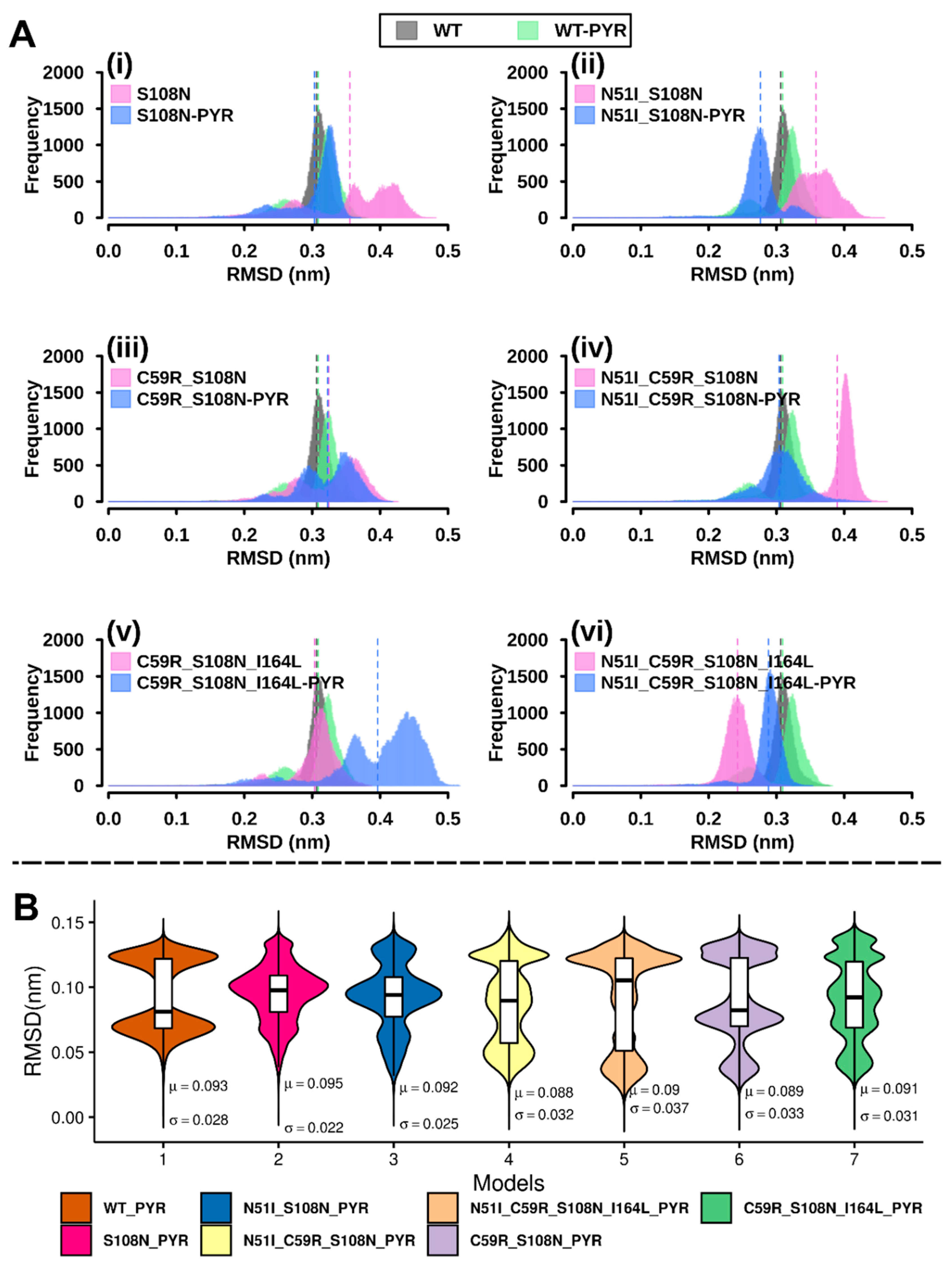
2.2.1. RMSD Analysis
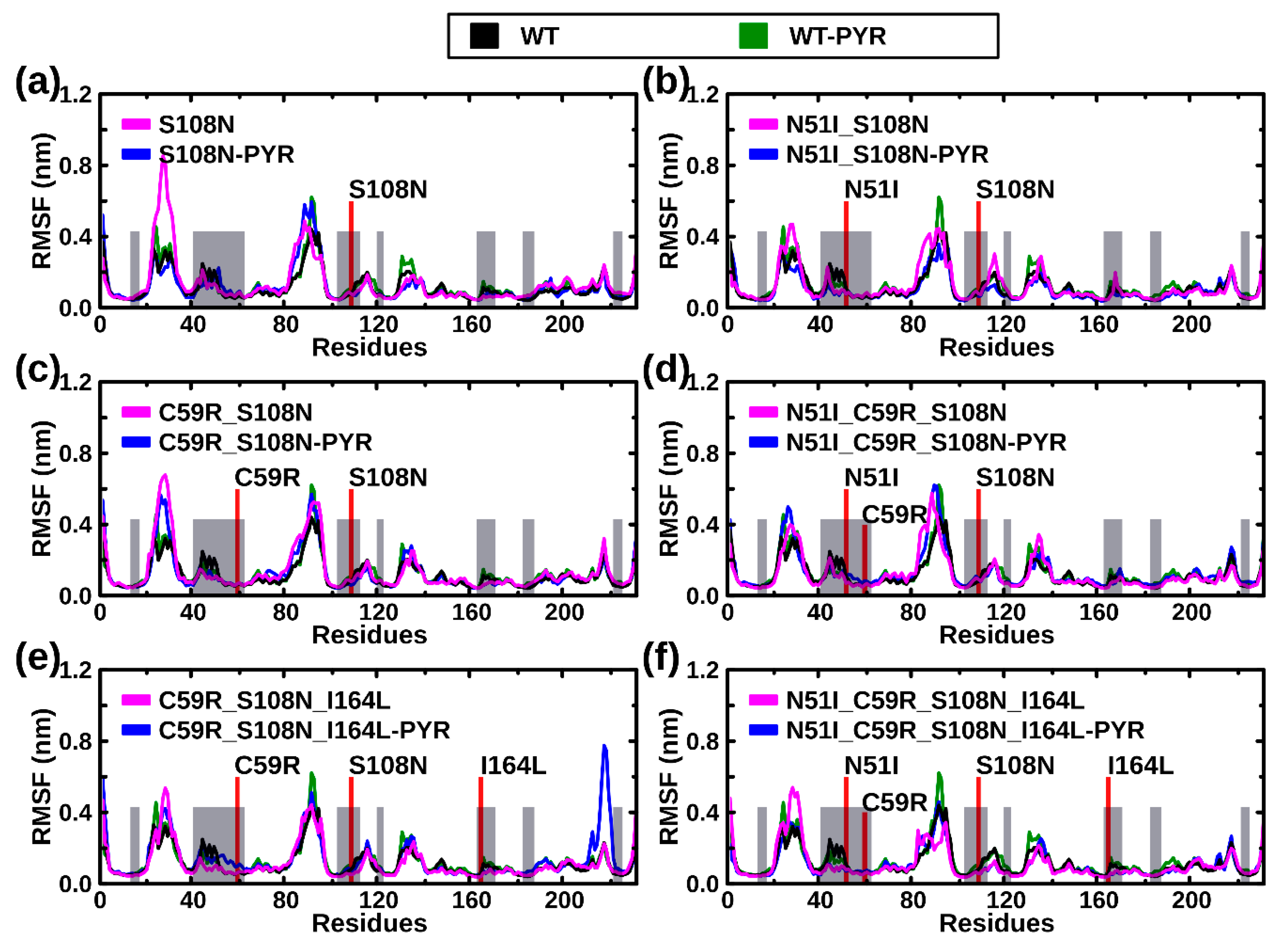
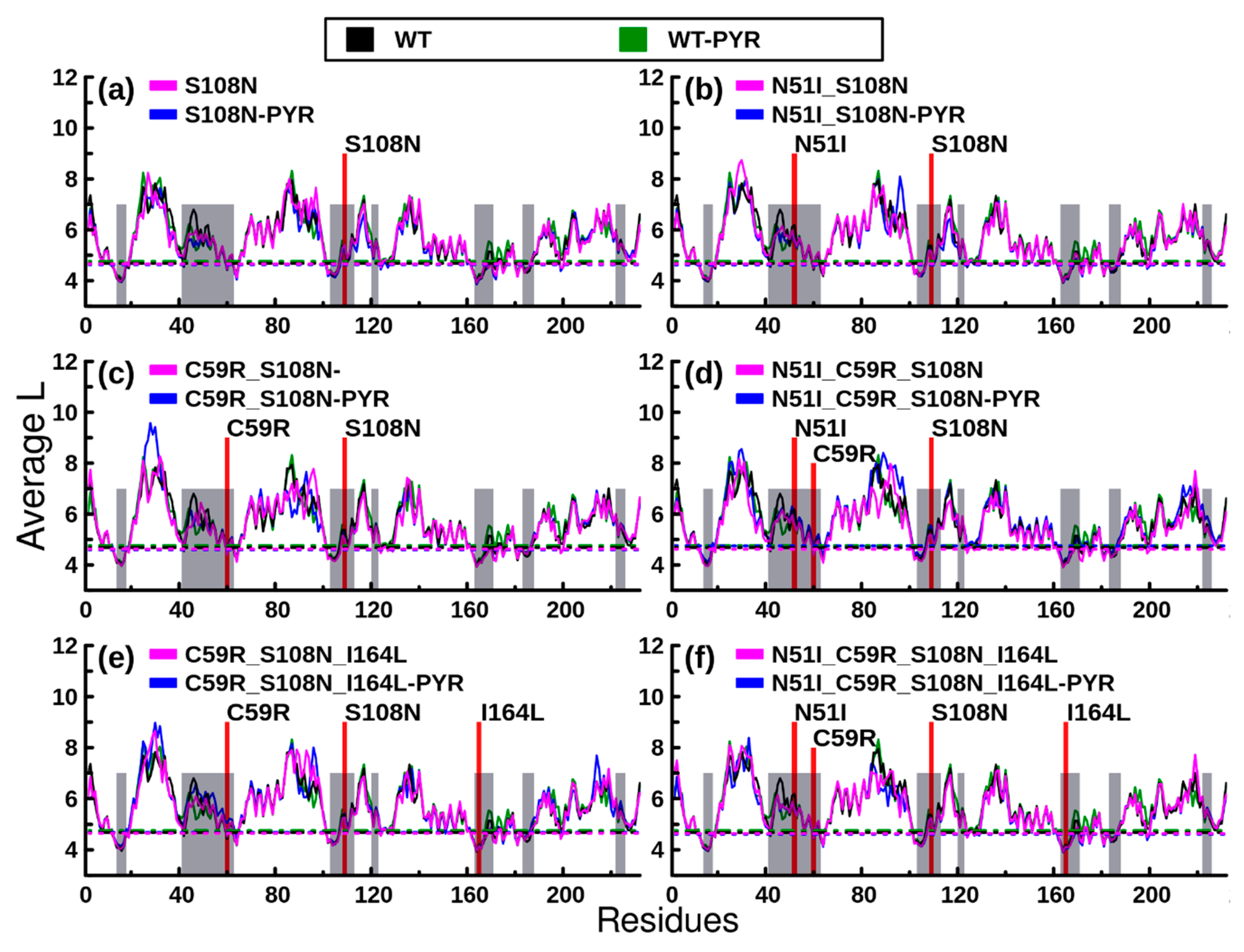
2.2.2. RMSF Analysis
2.2.3. Rg Analysis
2.2.4. Mutations Moderately Modulated Conformational Dynamics
2.2.5. Mutations Weakened the Binding Affinity of Pyrimethamine to DHFR
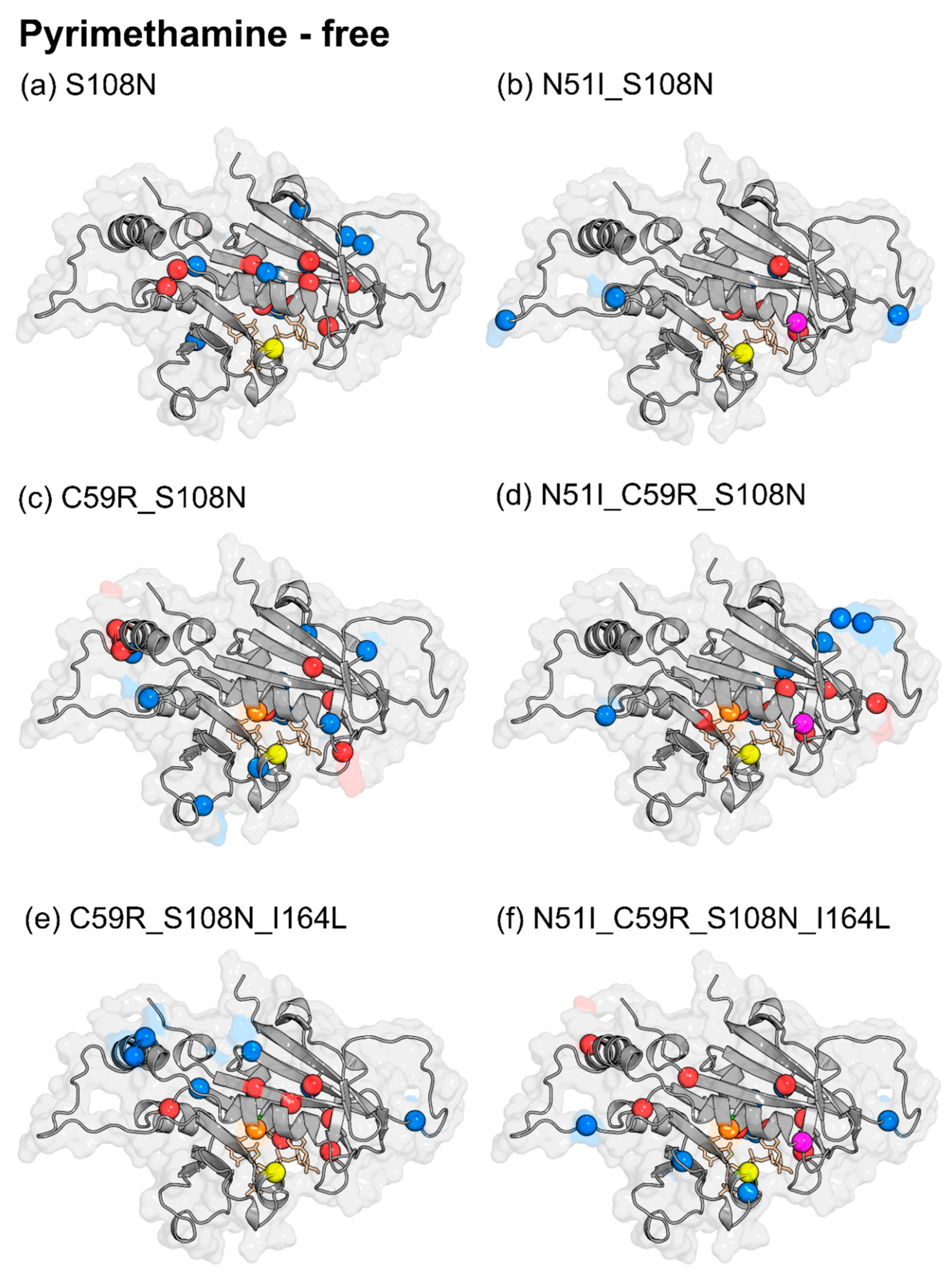
2.3. Differences in Intra-Protein Communication Patterns due to Mutations and Ligand Binding were Observed
2.3.1. Dynamic Residue Network Analysis
2.3.2. Mutation-Induced Changes in PfDHFR Intra-Protein Communication did not Directly Relate to Pyrimethamine Resistance
2.3.3. Pyrimethamine Binding Confers Unique Residue Communication Changes Across Different Mutants
3. Materials and Methods
3.1. Homology Modeling
3.2. Molecular Docking
3.3. Molecular Dynamics
3.3.1. Trajectory Analysis
3.3.2. Thermodynamic Assessment
3.3.3. Essential Dynamics
3.3.4. Dynamic Residue Network Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miller, L.H.; Baruch, D.I.; Marsh, K.; Doumbo, O.K. The pathogenic basis of malaria. Nature 2002, 415, 673–679. [Google Scholar] [CrossRef]
- WHO. World Malaria Report 2018; World Health Organization: Geneva, Switzerland, 2018; ISBN 9789241564106. [Google Scholar]
- Bruce Chwatt, L.J. Malaria and pregnancy. Br. Med. J. 1983, 286, 1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulman, C.E. Intermittent sulphadoxine-pyrimethamine to prevent severe anaemia secondary to malaria in pregnancy: A randomised placebo-controlled trial. Lancet 1999, 353, 632–636. [Google Scholar] [CrossRef]
- Falade, C.O.; Yusuf, B.O.; Fadero, F.F.; Mokuolu, O.A.; Hamer, D.H.; Salako, L.A. Intermittent preventive treatment with sulphadoxine-pyrimethamine is effective in preventing maternal and placental malaria in Ibadan, south-western Nigeria. Malar. J. 2007, 88. [Google Scholar] [CrossRef] [Green Version]
- Dicko, A.; Sagara, I.; Sissoko, M.S.; Guindo, O.; Diallo, A.I.; Kone, M.; Toure, O.B.; Sacko, M.; Doumbo, O.K. Impact of intermittent preventive treatment with sulphadoxine-pyrimethamine targeting the transmission season on the incidence of clinical malaria in children in Mali. Malar. J. 2008, 7, 123. [Google Scholar] [CrossRef] [Green Version]
- Yuthavong, Y.; Tarnchompoo, B.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Charman, S.A.; McLennan, D.N.; White, K.L.; Vivas, L.; Bongard, E.; et al. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc. Natl. Acad. Sci. USA 2012, 109. [Google Scholar] [CrossRef] [Green Version]
- Yuthavong, Y.; Yuvaniyama, J.; Chitnumsub, P.; Vanichtanankul, J.; Chusacultanachai, S.; Tarnchompoo, B.; Vilaivan, T.; Kamchonwongpaisan, S. Malarial (Plasmodium falciparum) dihydrofolate reductase-thymidylate synthase: Structural basis for antifolate resistance and development of effective inhibitors. Parasitology 2005, 130, 249–259. [Google Scholar] [CrossRef]
- Abbat, S.; Jain, V.; Bharatam, P.V. Origins of the specificity of inhibitor P218 toward wild-type and mutant PfDHFR: A molecular dynamics analysis. J. Biomol. Struct. Dyn. 2015, 33, 1319–1327. [Google Scholar] [CrossRef]
- Roper, C.; Pearce, R.; Nair, S.; Sharp, B.; Nosten, F.; Anderson, T. Intercontinental spread of pyrimethamine-resistant malaria. Science 2004, 305, 1124. [Google Scholar] [CrossRef]
- Gregson, A.; Plowe, C.V. Mechanisms of resistance of malaria parasites to antifolates. Pharmacol. Rev. 2005, 57, 117–145. [Google Scholar] [CrossRef] [Green Version]
- Bloland, P.B. Drug Resistance in Malaria (WHO/CDS/CSR/DRS/2001.4). 2001. Available online: http//whqlibdoc.who.int/hq/2001/WHO_CDS_CSR_DRS_2001.4.pdf (accessed on 20 October 2019).
- Antony, H.A.; Parija, S.C. Antimalarial drug resistance: An overview. Trop. Parasitol. 2016, 6, 30. [Google Scholar] [PubMed] [Green Version]
- Le Bras, J.; Durand, R. The mechanisms of resistance to antimalarial drugs in Plasmodium falciparum. Fundam. Clin. Pharmacol. 2003, 17, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Sirawaraporn, W.; Sathitkul, T.; Sirawaraporn, R.; Yuthavong, Y.; Santi, D.V. Antifolate-resistant mutants of Plasmodium falciparum dihydrofolate reductase. Proc. Natl. Acad. Sci. USA 1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basco, L.K.; de Pécoulas, P.E.; Wilson, C.M.; Le Bras, J.; Mazabraud, A. Point mutations in the dihydrofolate reductase-thymidylate synthase gene and pyrimethamine and cycloguanil resistance in Plasmodium falciparum. Mol. Biochem. 1995, 69, 135–138. [Google Scholar] [CrossRef]
- Choowongkomon, K.; Theppabutr, S.; Songtawee, N.; Day, N.P.J.; White, N.J.; Woodrow, C.J.; Imwong, M. Computational analysis of binding between malarial dihydrofolate reductases and anti-folates. Malar. J. 2010, 9, 65. [Google Scholar] [CrossRef] [Green Version]
- Mokmak, W.; Chunsrivirot, S.; Hannongbua, S.; Yuthavong, Y.; Tongsima, S.; Kamchonwongpaisan, S. Molecular dynamics of interactions between rigid and flexible antifolates and dihydrofolate reductase from pyrimethamine-sensitive and pyrimethamine-resistant plasmodium falciparum. Chem. Biol. Drug Des. 2014, 84, 450–461. [Google Scholar] [CrossRef]
- Tosso, R.D.; Andujar, S.A.; Gutierrez, L.; Angelina, E.; Rodríguez, R.; Nogueras, M.; Baldoni, H.; Suvire, F.D.; Cobo, J.; Enriz, R.D. Molecular modeling study of dihydrofolate reductase inhibitors. Molecular dynamics simulations, quantum mechanical calculations, and experimental corroboration. J. Chem. Inf. Modeling 2013, 53, 2018–2032. [Google Scholar] [CrossRef]
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Carlton, J.M.; Pain, A.; Nelson, K.E.; Bowman, S.; et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002, 419, 498–511. [Google Scholar] [CrossRef]
- Chaianantakul, N.; Sirawaraporn, R.; Sirawaraporn, W. Insights into the role of the junctional region of Plasmodium falciparum dihydrofolate reductase-thymidylate synthase. Malar. J. 2013, 12, 91. [Google Scholar] [CrossRef] [Green Version]
- Sanyanga, T.A.; Nizami, B.; Bishop, Ö.T. Mechanism of Action of Non-Synonymous Single Nucleotide Variations Associated with α-Carbonic Anhydrase II Deficiency. Molecules 2019, 24, 3987. [Google Scholar] [CrossRef] [Green Version]
- Amusengeri, A.; Astl, L.; Lobb, K.; Verkhivker, G.M.; Bishop, Ö.T. Establishing computational approaches towards identifying malarial allosteric modulators: A case study of plasmodium falciparum hsp70s. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penkler, D.; Bishop, O.T. Modulation of Human Hsp90α Conformational Dynamics by Allosteric Ligand Interaction at the C-Terminal Domain. Sci. Rep. 2018, 9, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.-J.; Nussinov, R. The free energy landscape in translational science: How can somatic mutations result in constitutive oncogenic activation? Phys. Chem. Chem. Phys. 2014, 16, 6332. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Cocco, L.; Roth, B.; Temple, C.; Montgomery, J.A.; London, R.E.; Blakley, R.L. Protonated state of methotrexate, trimethoprim, and pyrimethamine bound to dihydrofolate reductase. Arch. Biochem. Biophys. 1983, 226, 567–577. [Google Scholar] [CrossRef]
- Abdizadeh, H.; Tamer, Y.T.; Acar, O.; Toprak, E.; Atilgan, A.R.; Atilgan, C. Increased substrate affinity in the Escherichia coli L28R dihydrofolate reductase mutant causes trimethoprim resistance. Phys. Chem. Chem. Phys. 2017. [Google Scholar] [CrossRef]
- Bhakat, S.; Martin, A.J.M.; Soliman, M.E.S. An integrated molecular dynamics, principal component analysis and residue interaction network approach reveals the impact of M184V mutation on HIV reverse transcriptase resistance to lamivudine. Mol. Biosyst. 2014, 10, 2215–2228. [Google Scholar] [CrossRef]
- Amusengeri, A.; Bishop, Ö.T. Discorhabdin A South African natural compound, for Hsp72 and Hsc70 allosteric modulation: Combined study of molecular modeling and dynamic residue network analysis. Molecules 2019, 24, 188. [Google Scholar] [CrossRef] [Green Version]
- Doshi, U.; Holliday, M.J.; Eisenmesser, E.Z.; Hamelberg, D. Dynamical network of residue-residue contacts reveals coupled allosteric effects in recognition, catalysis, and mutation. Proc. Natl. Acad. Sci. USA 2016, 113, 4735–4740. [Google Scholar] [CrossRef] [Green Version]
- Penkler, D.; Atilgan, C.; Tastan Bishop, Ö. Allosteric Modulation of Human Hsp90α Conformational Dynamics. J. Chem. Inf. Modeling 2018, 58, 383–404. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.K.; Sheik Amamuddy, O.; Tastan Bishop, Ö. Structure-Based Analysis of Single Nucleotide Variants in the Renin-Angiotensinogen Complex. Glob. Heart 2017, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Yuvaniyama, J.; Chitnumsub, P.; Kamchonwongpaisan, S.; Vanichtanankul, J.; Sirawaraporn, W.; Taylor, P.; Walkinshaw, M.D.; Yuthavong, Y. Insights into antifolate resistance from malarial DHFR-TS structures. Nat. Struct. Biol. 2003, 10, 357–365. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, R.H.; Lilien, R.H.; Donald, B.R.; Stroud, R.M.; Anderson, A.C. Phylogenetic Classification of Protozoa Based on the Structure of the Linker Domain in the Bifunctional Enzyme, Dihydrofolate Reductase-Thymidylate Synthase. J. Biol. Chem. 2003, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkhivker, G.M. Biophysical simulations and structure-based modeling of residue interaction networks in the tumor suppressor proteins reveal functional role of cancer mutation hotspots in molecular communication. Biochim. Biophys. Acta (BBA) Gen. Subj. 2019, 1863, 210–225. [Google Scholar] [CrossRef]
- Brown, D.; Penkler, D.; Sheik Amamuddy, O.; Ross, C.; Atilgan, A.R.; Atilgan, C.; Tastan Bishop, Ö. MD-TASK: A software suite for analyzing molecular dynamics trajectories. Bioinformatics 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šali, A. MODELLER: A Program for Protein Structure Modeling Release 9.12, r9480. Rockefeller Univ. 2013. [Google Scholar]
- Hatherley, R.; Brown, D.K.; Glenister, M.; Tastan Bishop, Ö. PRIMO: An Interactive Homology Modeling Pipeline. PLoS ONE 2016, 11, e0166698. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.; Kim, B.-H.; Grishin, N.V. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B.; De Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: φ,ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Nature 1997, 356, 83–85. [Google Scholar]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BIOVA. BIOVIA Discovery Studio|Predictive Modeling & Science Simulation Software App. 2015. Available online: http://accelrys.com/products/collaborative-science/biovia-discovery-studio/ (accessed on 27 May 2016).
- DeLano, W.L. The PyMOL Molecular Graphics System, Version 1.7 Schrödinger, LLC. 2016. Available online: https://www.pymol.org/ (accessed on 16 May 2016).
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Modeling 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A Point-Charge Force Field for Molecular Mechanics Simulations of Proteins Based on Condensed-Phase Quantum Mechanical Calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed. 1999. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa -A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Modeling 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | ΔEvdW | ΔEelec | ΔGpolar | ΔGnonpolar | ΔG binding (kJ mol−1) |
|---|---|---|---|---|---|
| WT | −145.40 (0.20) | −5.97 (0.06) | 38.76 (0.10) | −14.58 (0.02) | −127.20 (0.21) |
| S108N | −136.80 (0.19) | −7.32 (0.06) | 50.06 (0.15) | −14.21 (0.02) | −108.26 (0.21) |
| N51I_S108N | −124.09 (0.19) | −6.63 (0.06) | 32.05 (0.11) | −14.46 (0.02) | −113.12 (0.20) |
| C59R_S108N | −127.74 (0.21) | −5.28 (0.07) | 62.50 (0.16) | −13.90 (0.02) | −84.40 (0.23) |
| N51I_ C59R_S108N | −130.53 (0.24) | −4.29 (0.06) | 47.76 (0.18) | −13.82 (0.02) | −100.87 (0.26) |
| C59R_S108N_I164L | −105.48 (0.26) | −2.59 (0.06) | 28.69 (0.11) | −13.40 (0.02) | −92.77 (0.25) |
| N51I_C59R_S108N_I164L | −116.49 (0.18) | −6.98 (0.08) | 53.07 (0.21) | −14.49 (0.02) | −84.90 (0.25) |
| Effect of Mutation (pyrimethamine-free: WT-free less mutant-free) | ||
|---|---|---|
| Protein | ΔBC | Residues |
| S108N | ↑ | Ala13, Ala16, Cys18, Gly41, Tyr158, Tyr159, Gly165, Ser167, Thr185 |
| ↓ | Ile14, Tyr35, Thr36, Ile143, Lys160, Val168, Gln171, Pro198, Asn201 | |
| N51I_S108N | ↑ | Gly41, Gly165, Ser167, Thr185 |
| ↓ | Lys23, Asp91, Asn157, Gln171, Pro198 | |
| C59R_S108N | ↑ | Lys49, Cys78, Lys79, Gly165, Thr185, Phe196 |
| ↓ | Thr36, Asn51, Asp81, Arg106, Lys132, Asn157, Val168, Gln171, Ile200 | |
| N51I_C59R_S108N | ↑ | Ala16, Cys18, Glu21, Gly41, Val103, Gly165, Ser167 |
| ↓ | Phe32, Asn33, Tyr35, Lys96, Val168, Gln171, Pro198 | |
| C59R_S108N_I164L | ↑ | Ala13, Cys15, Gly41, Tyr158, Gly166, Thr185, Phe196 |
| ↓ | Ser22, Glu71, Lys72, Lys160, Gln171, Lys181, Pro198 | |
| N51I_C59R_S108N_I164L | ↑ | Ile11, Gly41, Lys79, Tyr158, Gly165, Ser167, Thr185, Phe196 |
| ↓ | Ser22, Ser95, Ile112, Asn124, Val168, Gln171, Pro198 | |
| Effect of Mutation (pyrimethamine-bound: WT-PYR less mutant-PYR) | ||
| S108N | ↑ | Cys17, Gly26, Leu46, Cys78, Gly105, Trp109, Gly165, Gly166 |
| ↓ | Lys97, Val101, Ser167, Val168, Tyr170, Gln171 | |
| N51I_S108N | ↑ | Cys17, Leu46, Pro47, Met55, Gly105, Gly166, Leu174 |
| ↓ | Glu21, Gly41, Tyr158, Tyr159, Ser167, Tyr170, Gln171 | |
| C59R_S108N | ↑ | Ile11, Cys17, Val20, Leu46, Pro47, Gly105, Trp109, Gly165 |
| ↓ | Asn24, Phe32, Thr36, Lys97, Ser167, Val168, Tyr170, Gln171 | |
| N51I_C59R_S108N | ↑ | Cys17, Leu46, Pro47, Gly105, Trp109 |
| ↓ | Cys15, Tyr35, Gly41, Ser167, Val168, Lys180, Asn201 | |
| C59R_S108N_I164L | ↑ | Ala16, Cys17, Leu46, Pro47, Met55, Arg59, Gly105, Trp109, Gly165, Lys181 |
| ↓ | Ser22, Ser167, Val168, Tyr170, Gln171, Pro198, Thr220 | |
| N51I_C59R_S108N_I164L | ↑ | Cys17, Val20, Leu46, Pro47, Gly105, Trp109, Gly165 |
| ↓ | Glu21, Gly41, Asn90, Ser167, Val168, Tyr170, Gln171, Lys180, Pro198 | |
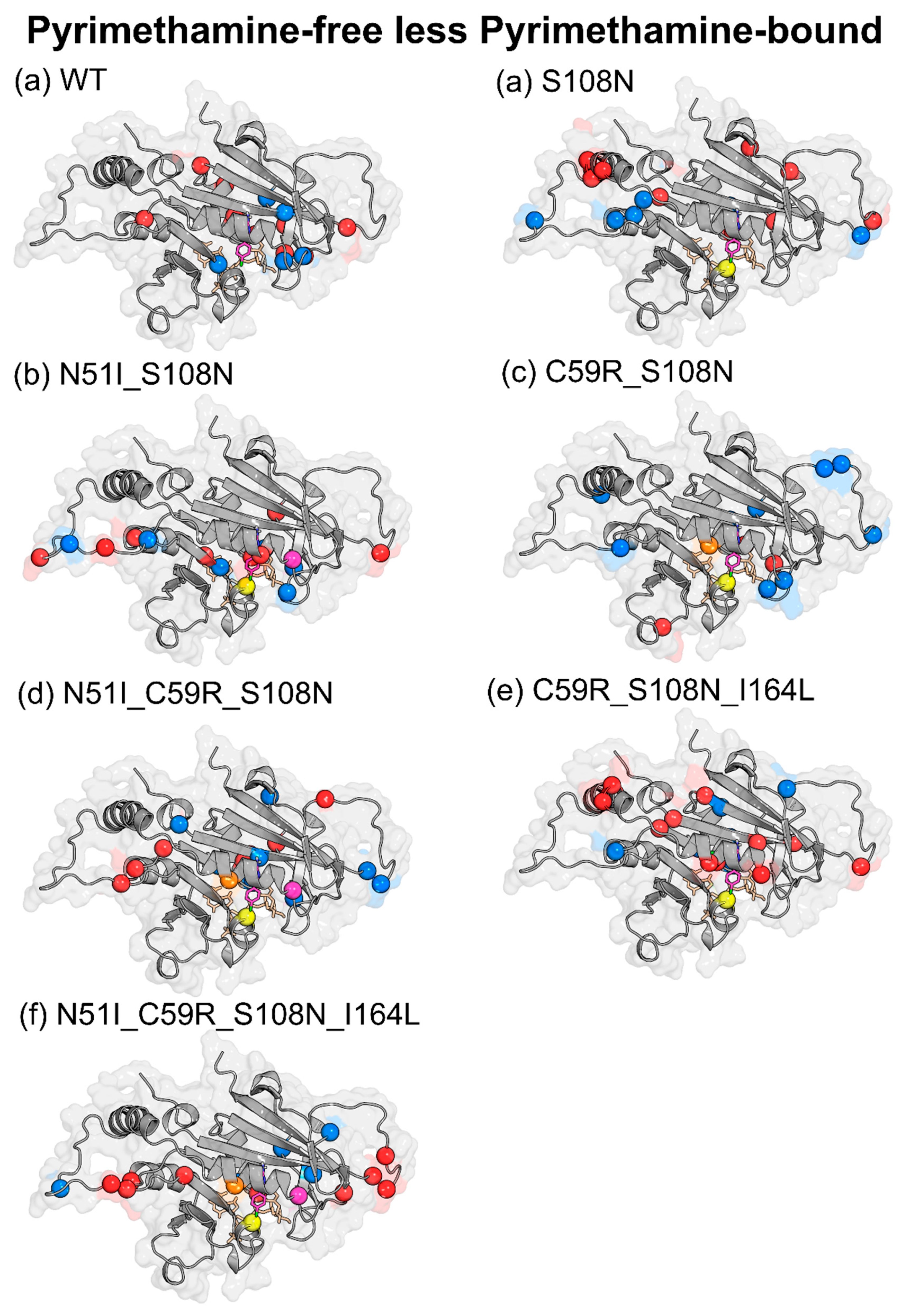
| Effect of Pyrimethamine (PYR-free less PYR-bound) | ||
| WT | ↑ | Glu21, Gly41, Tyr158, Ser167, Tyr170, Glu175, Lys180, Asp194, Phe196 |
| ↓ | Cys17, Leu46, Pro47, Gly105, Pro198 | |
| S108N | ↑ | Asn24, Tyr35, Glu71, Cys78, Lys79, Lys160, Val168, Phe196, AS201 |
| ↓ | Lys23, Val89, Asn157, Tyr158, Tyr159, Gln171 | |
| N51I_S108N | ↑ | Lys23, Met55, Asp91, Ser95, Val103, Asn157, Gly166, Pro198 |
| ↓ | Gly41, Val45, Pro93, Leu98, Met104, Ser167 | |
| C59R_S108N | ↑ | Gly41, Asp81, Met81, Lys132, Ser167 |
| ↓ | Asn24, Val31, Phe32, Val45, Lys49, Ser81, Lys81, Lys97, Gln171, Pro198 | |
| N51I_C59R_S108N | ↑ | Asn33, Lys96, Asn157, Tyr159, Tyr170, Gln171, Pro198 |
| ↓ | Asp10, Cys15, Ser22, Lys23, Gly41, Ile163, Val168, Asn201 | |
| C59R_S108N_I164L | ↑ | Ile11, Ala16, Cys18, Lys23, Ser52, Arg59, Glu71, Lys72, Lys160, Gly165, Lys181 |
| ↓ | Asn34, Asn157, Ile163, Val168, Gln171, Ile182 | |
| N51I_C59R_S108N_I164L | ↑ | Ser22, Lys23, Lys27, Ser95, Lys96, Asn100, Lys155, Gly166, Tyr191 |
| ↓ | Thr36, Gly39, Asn90, Leu164, Pro198 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amusengeri, A.; Tata, R.B.; Tastan Bishop, Ö. Understanding the Pyrimethamine Drug Resistance Mechanism via Combined Molecular Dynamics and Dynamic Residue Network Analysis. Molecules 2020, 25, 904. https://doi.org/10.3390/molecules25040904
Amusengeri A, Tata RB, Tastan Bishop Ö. Understanding the Pyrimethamine Drug Resistance Mechanism via Combined Molecular Dynamics and Dynamic Residue Network Analysis. Molecules. 2020; 25(4):904. https://doi.org/10.3390/molecules25040904
Chicago/Turabian StyleAmusengeri, Arnold, Rolland Bantar Tata, and Özlem Tastan Bishop. 2020. "Understanding the Pyrimethamine Drug Resistance Mechanism via Combined Molecular Dynamics and Dynamic Residue Network Analysis" Molecules 25, no. 4: 904. https://doi.org/10.3390/molecules25040904
APA StyleAmusengeri, A., Tata, R. B., & Tastan Bishop, Ö. (2020). Understanding the Pyrimethamine Drug Resistance Mechanism via Combined Molecular Dynamics and Dynamic Residue Network Analysis. Molecules, 25(4), 904. https://doi.org/10.3390/molecules25040904