3.1. Chemistry
3.1.1. General Information
Commercially available chemicals were of reagent grade and used as received. Purification of the compounds was performed by column chromatography on silica gel 60 M (0.040–0.063 mm, E. Merck, Darmstadt, Germany). Thin layer chromatography (TLC), using silica gel plates (Kieselgel 60F254, E. Merck), was used to monitor reaction progress. A B-540 Melting Point apparatus (Büchi, New Castle, DE, USA) was used to measure melting points. The 1H-NMR and 13C-NMR spectra, in DMSO-d6 and CDCl3, were recorded at the Department of Chemistry, University of Warsaw, using an AVANCE III HD 300 MHz spectrometer (Bruker, Billerica, MA, USA); shift values in parts per million are relative to the SiMe4 internal reference. The resonance assignments were based on peak multiplicity and peak integration of recorded spectra, Multiplets were assigned as bs (broad singlet), s (singlet), d (doublet), t (triplet) dd (doublet of doublet), ddd (doublet of doublet of doublet) and m (multiplet). An LTQ Orbitrap Velos instrument (Thermo Scientific, Waltham, MA, USA) located at the Mass Spectrometry Laboratory of the Institute of Biochemistry and Biophysics PAS (Warsaw, Poland) was used to record high resolution mass spectra. A 6200 FT/IR spectrometer (Jasco, Easton, MD, USA) at the Laboratory of Optical Spectroscopy (Institute of Organic Chemistry PAS, Warsaw, Poland) was used to record IR spectra.
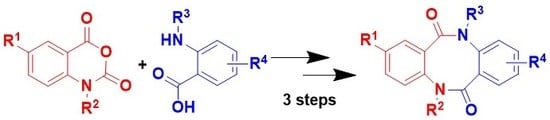
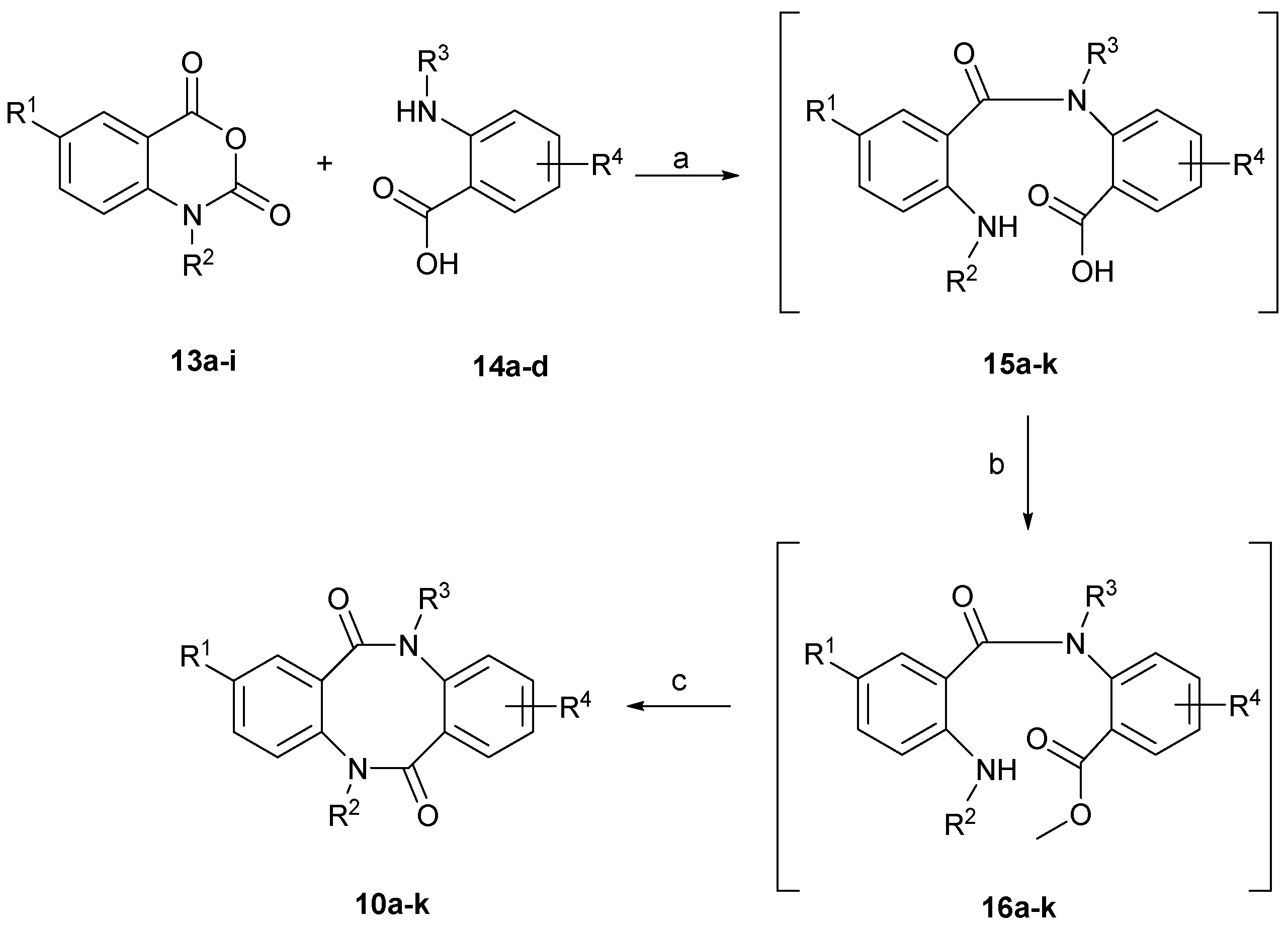
3.1.2. General Procedure for the Synthesis of 2-(2-aminobenzamido)benzoic acids 15a–k
A suspension of 1H-benzo[d][1,3]oxazine-2,4-dione 13a–i (1 equiv.), 2-aminobenzoic acid 14a–d (1 equiv.) and sodium hydroxide (1 equiv.) in water (10 mL/mmol) was heated at 80 °C for 30 min until the evolution of carbon dioxide had ceased and a clear solution had formed. After cooling the reaction mixture, the obtained solution was diluted with water, and the crude product was precipitated by addition of glacial acetic acid, filtered and dried under vacuum. In cases of N-alkyl 1H-benzo[d][1,3]oxazine-2,4-diones where a gummy-like residue was formed, the crude product was extracted with ethyl acetate, the organic phase was washed with brine and dried over anhydrous magnesium sulfate. Evaporation of the solvent left a glass-like residue which was used in next step without further purification.
3.1.3. General Procedure for the Synthesis of 2-(2-aminobenzamido)benzoic acids methyl esters 16a–k
2-(2-Aminobenzamido)benzoic acids 15a–k were dissolved in methanol (ca. 10 mL/mmol) then, concentrated sulfuric acid was added (0.5 mL/mmol) (caution: exothermic). The resulted solution was refluxed for 72 h. The excess of methanol was evaporated and the resulting residue was added to water. The pH was adjusted to 8 by addition of NaOH and the crude products were extracted with ethyl acetate. The combined organic layers were washed with 1 N sodium hydroxide, water and brine, and then dried over anhydrous magnesium sulfate. Evaporation of solvent yielded a dark residue of crude methyl esters 16a–k, which were used in the next step without further purification.
3.1.4. General Procedure for the Synthesis of dibenzo[b,f][1,5]diazocine-6,12(5H,11H)-diones 10a–k from 2-(2-aminobenzamido)benzoic acids methyl esters 16a–k
2-(2-Aminobenzamido)benzoic acids methyl esters 16a–k were dissolved in the anhydrous THF (20 mL/mmol) then 60% sodium hydride in mineral oil (2 equiv.) was added and the resulted solution was refluxed for 18 h. The excess of THF was evaporated, the obtained residue was poured into 1 N HCl and the crude product was extracted with ethyl acetate. The combined organic layers were washed with 1 N HCl, water and brine, then dried over anhydrous magnesium sulfate. Crude products were purified by column chromatography using hexane/EtOAc 1:1 then 2:8 v/v as eluent.
2-Chlorodibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dione (10a): Yield 33%, colourless crystals, m.p. 275–276 °C, R.f. = 0.40 (hexane:ethyl acetate 2:8 v/v). 1H-NMR (DMSO-d6) δ 10.33 (s, 1H, NH), 10.25 (s, 1H, NH), 7.45–7.21 (m, 5H, HAr), 7.11 (d, 1H, J = 2.4 Hz, HAr), 7.09 (d, 1H, J = 1.8 Hz, HAr); 13C-NMR (DMSO-d6) δ 169.1, 167.6, 135.3, 134.4, 133.8, 133.3, 131.4, 130.7, 130.4, 128.2, 127.7, 127.6, 127.5, 125.9; IR (KBr): cm−1 3183, 3055, 2900, 1658, 1601, 1578, 1484, 1415, 1366, 1261, 1229, 1145, 1111; HRMS (ESI): m/z [M+H]+ calcd for C14H10ClN2O2: 273.04253, 275.03958, found: 273.04202, 275.03904;
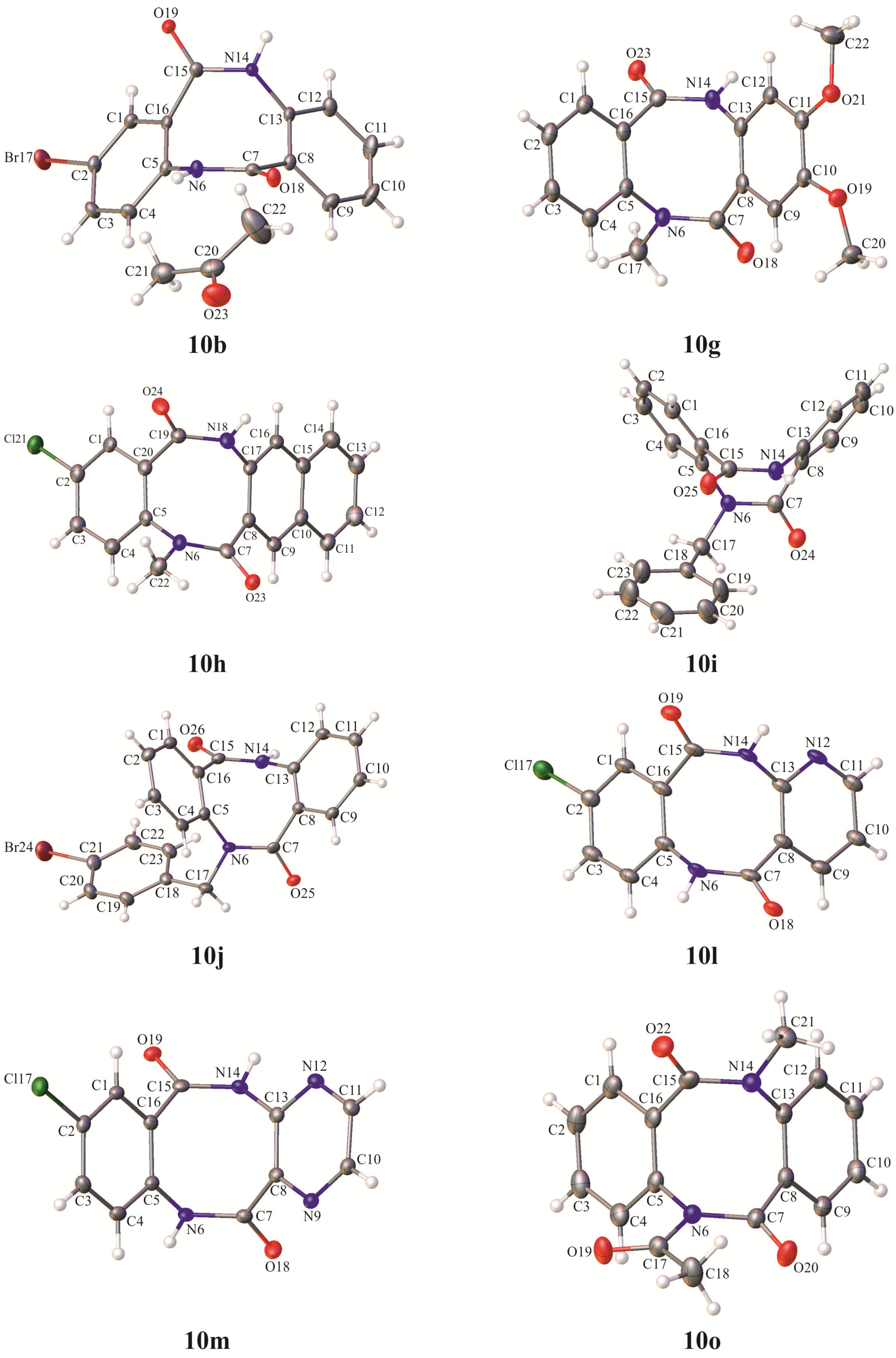
2-Bromodibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dione (10b): Yield 35%, colourless crystals, m.p. 283–284 °C, R.f. = 0.43 (hexane:ethyl acetate 2:8 v/v). 1H-NMR (DMSO-d6) δ 10.33 (s, 1H, NH), 10.24 (s, 1H, NH), 7.53 (dd, 1H, J = 2.4, 8.4 Hz, HAr), 7.49 (d, 1H, J = 2.4 Hz, HAr), 7.42–7.21 (m, 3H, HAr), 7.10 (d, 1H, J = 7.8 Hz, HAr), 7.03 (d, 1H, J = 8.4 Hz, HAr); 13C-NMR (DMSO-d6) δ 169.0, 167.5, 135.5, 134.4, 134.2, 133.3, 133.2, 130.7, 130.6, 128.2, 127.8, 127.5, 125.9, 119.6; IR (KBr): cm−1 3182, 3056, 2900, 1657, 1602, 1480, 1411, 1362, 1260, 1229, 1142, 1100; HRMS (ESI): m/z [M+H]+ calcd for C14H10BrN2O2: 316.99202, 318.98997, found: 316.99140, 318.98930;
8-Bromo-2,3-dimethoxydibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dione (10c): Yield 33%, colourless crystals, m.p. 195–196 °C, R.f. = 0.28 (ethyl acetate). 1H-NMR (DMSO-d6) δ 10.099 (s, 1H, NH), 10.057 (s, 1H, NH), 7.54 (dd, 1H, J = 2.4, 8.4 Hz, HAr), 7.46 (d, 1H, J = 2.4 Hz, HAr), 7.01 (d, 1H, J = 8.4 Hz, HAr), 6.81 (s, 1H, HAr), 6.65 (s, 1H, HAr), 3.72 (s, 3H, OCH3), 3.70 (s, 3H, OCH3); 13C-NMR (DMSO-d6) δ 169.0, 167.6, 150.0, 147.7, 135.6, 134.6, 133.2, 130.6, 127.85, 127.77, 124.9, 119.4, 110.4, 109.1, 55.70, 55.66; IR (KBr): cm−1 3499, 3185, 3064, 2934, 2848, 1678, 1646, 1607, 1517, 1470, 1401, 1342, 1264, 1224, 1176, 1108, 1078, 1027; HRMS (ESI): m/z [M+H]+ calcd for C16H14BrN2O4: 377.01315, 379.01110, found: 377.01246, 379.01037;
5-Methyldibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dione (10d): Yield 31%, colourless crystals, m.p. 258–259 °C, R.f. = 0.36 (hexane:ethyl acetate 2:8 v/v). 1H-NMR (DMSO-d6) δ 10.25 (s, 1H, NH), 7.45–7.15 (m, 7H, HAr), 7.07–6.98 (m, 1H, HAr), 3.33 (s, 1H, CH3, partially overlapped with H2O signal); 13C-NMR (DMSO-d6) δ 168.9, 167.4, 140.1, 134.7, 134.4, 133.3, 130.9, 130.2, 128.2, 127.9, 127.5, 127.1, 125.7, 125.4, 36.5; IR (KBr): cm−1 3257, 3067, 2979, 2934, 1991, 1938, 1836, 1673, 1623, 1599, 1575, 1470, 1415, 1386, 1351, 1302, 1260, 1226, 1185, 1161, 1141, 1082, 1034; HRMS (ESI): m/z [M+H]+ calcd for C15H13N2O2: 253.09715, found: 253.09682;
2-Bromo-11-methyldibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dione (10e): Yield 30%, colourless crystals, m.p. 247–248 °C, R.f. = 0.46 (hexane:ethyl acetate 2:8 v/v). 1H-NMR (DMSO-d6) δ 10.30 (s, 1H, NH), 7.53–7.25 (m, 6H, HAr), 7.00 (d, 1H, J = 8.4 Hz, HAr), 3.32 (s, 1H, CH3, partially overlapped with H2O signal); 13C-NMR (DMSO-d6) δ 186.7, 165.8, 139.7, 136.3, 134.2, 133.1, 132.9, 131.2, 130.3, 128.5, 127.6. 127.5, 125.8, 119.4, 36.5; IR (KBr): cm−1 3172, 3087, 2881, 1681, 1635, 1597, 1473, 1427, 1399, 1370, 1345, 1300, 1274, 1240, 1180, 1135, 1079, 1038, 1014; HRMS (ESI): m/z [M+H]+ calcd for C15H12BrN2O2: 331.00767, 333.00562, found: 331.00718, 333.00503;
11-Methyl-2-nitrodibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dione (10f): Yield 25%, colourless crystals, m.p. 225–226 °C, R.f. = 0.46 (hexane:ethyl acetate 2:8 v/v). 1H-NMR (DMSO-d6) δ 10.74 (s, 1H, NH), 8.18-8.09 (m, 2H, HAr), 7.50–7.41 (m, 2H, HAr), 7.36–7.26 (m, 3H, HAr), 3.37 (s, 1H, CH3, overlapped with H2O signal); 13C-NMR (DMSO-d6) δ 168.6, 165.3, 145.5, 140.5, 139.6, 134.9, 132.4, 131.4, 128.7, 127.8, 126.4, 125.9, 125.3, 123.6, 36.6; IR (KBr): cm−1 3177, 3056, 2907, 1946, 1712, 1676, 1628, 1528, 1485, 1335, 1260, 1239, 1178, 1137, 1089; HRMS (ESI): m/z [M+H]+ calcd for C15H12N3O4: 298.08223, found: 298.08160;
2,3-Dimethoxy-11-methyldibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dione (10g): Yield 29%, colourless crystals, m.p. 271–272 °C, R.f. = 0.23 (ethyl acetate). 1H-NMR (, DMSO-d6) δ 9.97 (s, 1H, NH), 7.46–7.37 (m, 2H, HAr), 7.37–7.27 (m, 3H, HAr), 6.77 (s, 1H, HAr), 6.59 (s, 1H, HAr), 3.68 (s, 3H, OCH3), 3.67 (s, 3H, OCH3), 3.30 (s, 1H, CH3); 13C-NMR (DMSO-d6) δ 168.9, 167.3, 149.6, 147.4, 140.5, 133.3, 130.9, 128.1, 129.0, 127.6, 126.1, 125.6, 110.2, 108.7, 55.65, 55.62, 36.6; IR (KBr): cm−1 3193, 3000, 2936, 2841, 1674, 1626, 1513, 1478, 1454, 1431, 1398, 1364, 1302, 1258, 1223, 1156, 1133, 1105, 1082, 1036, 1021; HRMS (ESI): m/z [M+H]+ calcd for C17H17N2O4: 313.11828, found: 313.11760;
2-Chloro-5-methylbenzo[b]naphtho[2,3-f][1,5]diazocine-6,14(5H,13H)-dione (10h): Yield 34%, beige crystals, m.p. 270–271 °C, R.f. = 0.56 (hexane:ethyl acetate 2:8 v/v). 1H-NMR (DMSO-d6) δ 10.59 (s, 1H, NH), 7.98–7.82 (m, 3H, HAr), 7.62 (s, 1H, HAr), 7.58–7.36 (m, 5H, HAr), 3.37 (s, 1H, CH3); 13C-NMR (DMSO-d6) δ 167.6, 167.3, 138.6, 135.6, 133.4, 133.0, 132.6, 131.8, 131.1, 131.0, 128.1, 128.0, 127.78, 127.76, 127.5, 127.1, 126.9, 123.6, 36.5; IR (KBr): cm−1 3157, 3117, 3052, 2965, 2924, 2887, 1939, 1913, 1839, 1768, 1670, 1630, 1594, 1475, 1420, 1351, 1302, 1274, 1228, 1164, 1129, 1106; HRMS (ESI): m/z [M+H]+ calcd for C19H14ClN2O2: 337.07383, 339.07088, found: 337.07313, 339.07024;
5-Benzyldibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dione (10i): Yield 29%, beige crystals, m.p. 212–213 °C, R.f. = 0.60 (hexane:ethyl acetate). 1H-NMR (DMSO-d6) δ 10.26 (s, 1H, NH), 7.37–7.18 (m, 12H, HAr), 7.05–7.00 (m, 1H, HAr), 5.32 (d, 1H, J = 15.0 Hz, CH2), 4.81 (d, 1H, J = 15.0 Hz, CH2); 13C-NMR (DMSO-d6) δ 168.9, 167.8, 138.9, 136.7, 134.8, 134.3, 133.9, 130.8, 130.3, 128.3, 128.2, 127.9, 127.84, 127.76, 127.3, 127.1, 125.9, 125.4, 52.4; IR (KBr): cm−1 3166, 3037, 2952, 2898, 1969, 1829, 1735, 1664, 1638, 1598, 1486, 1454, 1402, 1379, 1301, 1240, 1153, 1104, 1076, 1042; HRMS (ESI): m/z [M+H]+ calcd for C21H17N2O2: 329.12845, found: 329.12792;
5-(4-Bromobenzyl)dibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dione (10j): Yield 30%, colourless crystals, m.p. 222–223 °C, R.f. = 0.66 (hexane:ethyl acetate). 1H-NMR (DMSO-d6) δ 10.24 (s, 1H, NH), 7.51–7.15 (m, 12H, HAr), 7.06–6.98 (m, 1H, HAr), 5.35 (d, 1H, J = 15.0 Hz, CH2), 4.72 (d, 1H, J = 15.0 Hz, CH2); 13C- NMR (DMSO-d6) δ 168.6, 167.7, 138.6, 136.1, 134.8, 134.2, 133.9, 131.2, 130.9, 130.35, 130.32, 128.4, 127.8, 127.7, 127.1, 126.0, 125.4, 120.5, 51.7; IR (KBr): cm−1 3219, 3059, 2939, 1901, 1672, 1633, 1598, 1488, 1467, 1395, 1353, 1307, 1285, 1258, 1217, 1158, 1103, 1068, 1024, 1009; HRMS (ESI): m/z [M+H]+ calcd for C21H16BrN2O2: 407.03897, 409.03692, found: 407.03841, 409.03626;
2-Bromo-11-methyl-5-(naphthalen-1-ylmethyl)dibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dione (10k): Yield 18%, colourless crystals, m.p. 255–256 °C, R.f. = 0.40 (hexane:ethyl acetate 7:3 v/v). 1H-NMR (DMSO-d6) δ 8.18–8.06 (m, 1H, HAr), 7.98–7.88 (m, 1H, HAr), 7.82 (d, 1H, J = 8.1 Hz, HAr), 7.60–7.22 (m, 9H, HAr), 7.16–7.07 (m, 2H, HAr), 6.16 (d, 1H, J = 15.0 Hz, CH2), 4.98 (d, 1H, J = 15.0 Hz, CH2), 3.05 (s, 1H, CH3); 13C-NMR (DMSO-d6) δ 166.8, 164.5, 139.8, 137.3, 136.6, 133.7, 133.4, 133.3, 131.4, 131.3, 131.0, 130.0, 128.6, 128.5, 128.4, 128.3, 127.6, 127.4, 126.3, 125.8, 125.7, 125.0, 123.3, 120.5, 48.8, 35.8; IR (KBr): cm−1 3039, 3037, 3006, 2978, 2930, 2904, 2875, 1731, 1663, 1641, 1597, 1510, 1469, 1452, 1422, 1409, 1355, 1285, 1259, 1209, 1175, 1155, 1123, 1082, 1042, 1016; HRMS (ESI): m/z [M+H]+ calcd for C26H20BrN2O2: 471.07027, 473.06819, found: 471.07027, 473.06822;
3.1.5. General Procedure for the Synthesis of 8-chloropyrido[3,2-c][1,5] benzodiazocine-5,11- (6H,12H)-dione (10l) and 8-chloropyrazino[3,2-c][1,5]benzodiazocine- 6,12(5H,11H)-dione (10m)
Synthesis of 2-amino-N-sulfinylnicotinoyl chloride (19a) and 3-amino-N-sulfinyl-pyrazine-2-carbonyl chloride (19b).
2-Aminonicotinic acid (18a) or 3-amino-2-pyrazinecarboxylic acid (18b) was suspended in dry toluene (10 mL/mmol), then thionyl chloride (5 equiv.) was added dropwise. The obtained slurry was refluxed for 3 h, until clear solution was formed. The excess of solvent was evaporated, and the residue was co-evaporated with toluene to remove traces of thionyl chloride. Crude products 19a,b were used in the next step without further purification.
Synthesis of methyl 2-(2-aminonicotinamido)-5-chlorobenzoate (21a) and methyl 2-(2-aminopyrazine-3-carboxamido)-5-chlorobenzoate (21b).
2-Amino-N-sulfinylnicotinoyl chloride (19a) or 3-amino-N-sulfinylpyrazine-2-carbonyl chloride (19b) (1 equiv.) was dissolved in dry toluene (10 mL/mmol), then the solution of methyl 2-amino-5-chlorobenzoate (20) (1 equiv.) in toluene (10 mL/mmol) was added dropwise. The mixture was stirred at ambient temperature for 48 h, then the solvent was evaporated and the obtained residue was dissolved in ethyl acetate. The organic phase was washed two times with 1 N NaOH, water and brine, then dried over anhydrous magnesium sulfate. Evaporation of the solvent resulted in crude methyl esters 21a-b which were used in next step without further purification.
Cyclisation of methyl 2-(2-aminonicotinamido)-5-chlorobenzoate (21a) and methyl 2-(2-aminopyrazine-3-carboxamido)-5-chlorobenzoate (21b).
Methyl 2-(2-aminonicotinamido)-5-chlorobenzoate (21a, 1 equiv.) and methyl 2-(2-aminopyrazine-3-carboxamido)-5-chlorobenzoate (21b, 1 equiv.) were dissolved in anhydrous THF (20 mL/mmol) then 60% sodium hydride in mineral oil (2 equiv.) was added. The resulting solution was refluxed for 18 h. The excess of THF was evaporated, residue was poured into 1 N HCl and the crude product was extracted with ethyl acetate. The combined organic layers were washed with 1 N HCl, water and brine, then dried with anhydrous magnesium sulfate. Crude products 10l,m were purified by column chromatography using hexane/EtOAc 1:1 then 2:8 v/v as eluent.
8-Chloropyrido[3,2-c][1,5]benzodiazocine-5,11(6H,12H)-dione (10l): Yield 17%, colourless crystals, m.p. 308-309 °C, R.f. = 0.26 (hexane:ethyl acetate). 1H-NMR (DMSO-d6) δ 10.84 (s, 1H, NH), 10.46 (s, 1H, NH), 8.46 (dd, 1H, J = 1.8, 4.8 Hz, HAr), 7.84 (dd, 1H, J = 1.8, 7.5 Hz, HAr), 7.49–7.40 (m, 2H, HAr), 7.48 (dd, 1H, J = 4.8, 7.8 Hz, HAr), 7.18–7.11 (m, 1H, HAr); 13C-NMR (DMSO-d6) δ 167.7, 167.3, 150.6, 147.0, 138.1, 134.6, 133.6, 131.7, 130.8, 128.0, 127.90, 127.64, 123.1; IR (KBr): cm−1 3182, 3066, 2937, 2902, 1924, 1675, 1596, 1486, 1459, 1433, 1410, 1328, 1281, 1254, 1226, 1150, 1110; HRMS (ESI): m/z [M+H]+ calcd for C13H9ClN3O2: 274.03778, 276.03483, found: 274.0372, 276.03436;
8-Chloropyrazino[3,2-c][1,5]benzodiazocine-6,12(5H,11H)-dione (10m): Yield 19%, colourless crystals, m.p. 346–347 °C, R.f. = 0.35 (hexane:ethyl acetate). 1H-NMR (DMSO-d6) δ 11.24 (s, 1H, NH), 10.77 (s, 1H, NH), 8.65–8.50 (m, 2H, HAr), 7.55–7.40 (m, 2H, HAr), 7.30–7.18 (m, 2H, HAr); 13C-NMR (DMSO-d6) δ 167.1, 165.5, 144.9, 144.5, 144.1, 143.7, 134.1, 132.8, 132.2, 131.1, 128.2, 127.8; IR (KBr): cm−1 3249, 3164, 3057, 2961, 2891, 1928, 1685, 1571, 1537, 1484, 1458, 1406, 1343, 1276, 1236, 1211, 1150, 1133, 1108, 1064; HRMS (ESI): m/z [M+H]+ calcd for C12H8ClN4O2: 275.03303, 277.03008, found: 275.03247, 277.02957;
3.1.6. Synthesis of ethyl 2-(11-methyl-6,12-dioxo-11,12-dihydrodibenzo[b,f][1,5]diazocin- 5(6H)-yl)acetate (10n)
5-Methyldibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dione (10d, 252 mg, 1 mmol, 1 equiv.) was dissolved in anhydrous DMSO (5 mL), then 60% NaH dispersed in mineral oil (48 mg, 1.2 mmol, 1.2 equiv.) was added. The resulted suspension was stirred at room temperature for 30 min. until evolution of gas ceased. Then, ethyl bromoacetate (133 μL, 1.2 mmol, 1.2 equiv.) was added dropwise, and the resulting solution was stirred for 18 h at room temperature. The next day, the reaction mixture was poured into water (20 mL) and extracted with ethyl acetate (3 × 20 mL). Combined organic layers were washed with brine (1 × 20 mL) and dried over anhydrous magnesium sulfate, followed by evaporation under reduced pressure. Crude product was purified by column chromatography using hexane/EtOAc 1:1 then 2:8 v/v as eluent. Yield 78% (264 mg), colourless crystals, m.p. 125–126 °C, R.f. = 0.66 (hexane:ethyl acetate 2:8 v/v). 1H-NMR (DMSO-d6) δ 7.45-7.15 (m, 8H, HAr), 4.62 (d, 1H, J = 17.1 Hz, CH2), 4.47 (d, 1H, J = 17.1 Hz, CH2), 4.23–4.10 (m, 2H, CH2), 1.22 (t, 3H, J = 7.2 Hz, CH3); 13C-NMR (DMSO-d6) δ 168.3, 167.6, 166.9, 140.1, 139.0, 134.3, 133.2, 131.0, 130.8, 128.2, 128.1, 127.6, 127.2, 125.6, 125.2, 60.9, 51.1, 36.1, 14.0; IR (KBr): cm−1 3458, 3300, 3058, 2981, 2936, 1953, 1840, 1739, 1656, 1599, 1456, 1411, 1411, 1375, 1324, 1308, 1281, 1260, 1217, 1120, 1088, 1056, 1023; HRMS (ESI): m/z [M+H]+ calcd for C19H19N2O4: 339.13393, found: 339.13393;
3.1.7. Synthesis of 5-acetyl-11-methyldibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dione (10o)
The dispersion of 5-methyldibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dione (10d) (504 mg, 2 mmol, 1 equiv.) in acetic anhydride (10 mL) was refluxed for 3 h. The excess anhydride was evaporated under reduced pressure and the residue was crystallized from a mixture of cyclohexane and ethyl acetate (9:1) to give pure product. Yield 89% (524 mg), colourless crystals, m.p. 193–194 °C, R.f. = 0.74 (hexane:ethyl acetate 2:8 v/v). 1H-NMR (DMSO-d6) δ 7.46-7.19 (m, 8H, HAr), 3.34 (s, 1H, CH3), 2.61 (s, 1H, CH3); 13C-NMR (DMSO-d6) δ 171.5, 168.6, 166.7, 139.7, 135.4, 134.3, 133.8, 132.1, 130.3, 129.4, 129.0, 128.3, 127.8, 127.0, 126.2, 36.0, 26.7; IR (KBr): cm−1 3412, 3379, 3296, 3061, 2938, 1975, 1941, 1718, 1703, 1658, 1599, 1485, 1453, 1420, 1371, 1304, 1251, 1206, 1142, 1084, 1038, 1013; HRMS (ESI): m/z [M+H]+ calcd for C17H15N2O3: 295.10772, found: 295.10735;
3.1.8. Synthesis of 8-bromo-2,3-dimethoxydibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dithione (10p)
Bromo-2,3-dimethoxydibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dione (10c, 376 mg, 1 mmol, 1 equiv.) was dissolved in dry THF and then p-tolyl Davy-reagent (873 mg, 2 mmol, 2 equiv.) was added. The resulting suspension was refluxed for 18 h, then the reaction mixture was cooled to room temperature. The obtained clear, yellow solution was evaporated with small amount of chromatographic silica gel. The crude product was purified by column chromatography using hexane/EtOAc 9:1 then 7:3 v/v as eluent. Yield 58% (237 mg), yellow solid, m.p. 186–187 °C (decomposition), R.f. = 0.27 (hexane:ethyl acetate 7:3 v/v). 1H-NMR (DMSO-d6) δ 12.30 (s, 1H, NH), 12.28 (s, 1H, NH), 7.56–7.49 (m, 2H, HAr), 6.99 (d, 1H, J = 5.1 Hz, HAr), 6.88 (s, 1H, HAr), 6.64 (s, 1H, HAr), 3.73 (s, 3H, OMe), 3.70 (s, 3H, OMe); 13C-NMR (DMSO-d6) δ 200.2, 197.5, 149.8, 148.0, 141.2, 133.1, 132.9, 131.2, 131.1, 126.9, 126.6, 120.4, 111.4, 107.7, 55.9, 55.7; IR (KBr): cm−1 3439, 3248, 2925, 2644, 1602, 1506, 1403, 1345, 1265, 1227,1200, 1141, 1079, 1030; HRMS (ESI): m/z [M+H]+ calcd for C16H14BrN2O2S2: 408.96746, 410.96541, found: 408.96725, 410.96512;
3.1.9. General Procedure for the Synthesis of 1-Substituted 1H-benzo[d][1,3]oxazine-2,4-diones 13f–i
To a stirred solution of 6-chloro-1H-benzo[d][1,3]oxazine-2,4-dione (13a), 6-bromo-1H- benzo[d][1,3]oxazine-2,4-dione (13b) or 1H-benzo[d][1,3]oxazine-2,4-dione (13c) (1 equiv.) in DMSO (10 mL/mmol), 60% sodium hydride (1.5 equiv.) was added and the resulting suspension was stirred for 15 min. at room temperature, until the evolution of gas ceased. Then, the appropriate halide (1.5 equiv.): methyl iodide for 13a, 1-(bromomethyl)naphthalene for 13b or benzyl bromide and 4-bromobenzyl bromide for 13c, was added and the resulting mixture was stirred at room temperature for 18 h. Next day, the reaction mixture was poured into water (100 mL/10 mL DMSO) and extracted with ethyl acetate (3 × 100 mL/100 mL H2O). Combined organic layers were washed with brine (100 mL/300 mL EtOAc) and dried over anhydrous magnesium sulfate. Crude products were crystallized from mixture of ethyl acetate and hexane.
6-Chloro-1-methyl-1H-benzo[d][1,3]oxazine-2,4-dione (13f): Yield 62%, yellow crystals, m.p. 201–202 °C, R.f. = 0.33 (hexane:ethyl acetate 7:3). 1H-NMR (DMSO-d6) δ 7.94 (d, 1H, J = 2.4 Hz, HAr), 7.89 (dd, 1H, J = 2.4, 9.0 Hz, HAr), 7.48 (d, 1H, J = 9.0 Hz, HAr), 3.46 (s, 1H, Me); 13C-NMR (DMSO-d6) δ 158.0, 147.4, 141.1, 136.6, 127.9, 127.6, 117.1, 113.3, 31.9; HRMS (ESI): m/z [M+H]+ calcd for C9H7ClNO3: 212.01090, 214.00795, found: 212.01049, 214.00754;
1-Benzyl-1H-benzo[d][1,3]oxazine-2,4-dione (13g): Yield 57%, beige crystals, m.p. 141–142 °C, R.f. = 0.54 (hexane:ethyl acetate 7:3). 1H-NMR (DMSO-d6) δ 8.04 (dd, 1H, J = 1.5, 7.8 Hz, HAr), 7.73 (ddd, 1H, J = 1.5, 7.2, 8.7 Hz, HAr), 7.46–7.20 (m, 7H, HAr), 5.30 (s, 2H, CH2); 13C-NMR (DMSO-d6) δ 158.9, 148.3, 141.3, 137.0, 135.3, 129.5, 128.6, 127.4, 126.6, 123.7, 115.1, 112.1, 47.6; HRMS (ESI): m/z [M+H]+ calcd for C15H12NO3: 254.08116, found: 254.08104;
1-(4-Bromobenzyl)-1H-benzo[d][1,3]oxazine-2,4-dione (13h): Yield 62%, beige crystals, m.p. 182–183 °C, R.f. = 0.50 (hexane:ethyl acetate 7:3). 1H-NMR (DMSO-d6) δ 8.04 (dd, 1H, J = 1.5, 7.8 Hz, HAr), 7.74 (ddd, 1H, J = 1.5, 7.2, 8.7 Hz, HAr), 7.85–7.49 (m, 2H, HAr), 7.44–7.36 (m, 2H, HAr), 7.35–7.26 (m, 1H, HAr), 7.22 (dd, 1H, J = 8.4 Hz, HAr), 5.27 (s, 2H, CH2); 13C-NMR (DMSO-d6) δ 158.8, 148.3, 141.2, 137.0, 134.8, 131.5, 129.5, 129.0, 123.8, 120.5, 115.0, 112.2, 47.0; HRMS (ESI): m/z [M+H]+ calcd for C15H11BrNO3: 331.99168, 333.98964, found: 331.99138, 333.98930;
6-Bromo-1-(naphthalen-1-ylmethyl)-1H-benzo[d][1,3]oxazine-2,4-dione (13i): Yield 51%, white solid, m.p. 230–231 °C, R.f. = 0.74 (hexane:ethyl acetate 7:3). 1H-NMR (DMSO-d6) δ 8.26–8.16 (m, 1H, HAr), 8.15 (d, 1H, J = 2.4 Hz, HAr), 8.04–7.97 (m, 1H, HAr), 7.92–7.78 (m, 2H, HAr), 7.72–7.56 (m, 2H, HAr), 7.42–7.33 (m, 2H, HAr), 7.02 (d, 1H, J = 9.0 Hz, HAr), 5.73 (s, 2H, CH2); 13C-NMR (DMSO-d6) δ 157.9, 147.8, 140.8, 139.2, 133.3, 131.0, 129.9, 129.5, 128.7, 127.7, 126.5, 126.2, 125.4, 123.0, 122.2, 117.7, 115.3, 114.52, 114.48, 46.4; HRMS (ESI): m/z [M+H]+ calcd for C19H13BrNO3: 382.0073, 384.00529 found: 382.0645, 384.00443;
3.1.10. DCC-mediated Synthesis of 2-(2-aminophenyl)-4H-benzo[d][1,3]oxazin-4-one (12, R1 = R2 = H)
A suspension of 1H-benzo[d][1,3]oxazine-2,4-dione (13c, 489 mg, 3 mmol, 1 equiv.), 2-aminobenzoic acid (14a, 411 mg, 3 mmol, 1 equiv.), sodium hydroxide (120 mg, 3 mmol, 1 equiv.) in water (30 mL) was heated at 80 °C for 30 min. until the evolution of carbon dioxide ceased and clear solution was formed. After cooling, the obtained solution was diluted with water, crude product was precipitated by addition of glacial acetic acid and the resulting precipitate was dried under reduced pressure. The crude 2-(2-aminobenzamido)benzoic acid (11) was dissolved in 50 mL of DMF, then N,N’-dicyclohexylcarbodiimide (DCC) (681 mg, 3.3 mmol, 1.1 equiv.) was added and the reaction mixture was stirred at room temperature for 18 h. The next day, the obtained solution was poured into water (100 mL) and the product was extracted with ethyl acetate (3 × 100 mL). Combined organic layers were washed with brine (1 × 50 mL), dried over anhydrous magnesium sulfate followed by evaporation of volatiles under reduced pressure. Crude product was purified by column chromatography using hexane: ethyl acetate 9:1 v/v as eluent. yield 84% (600 mg), yellow solid, m.p. 171–172 °C, R.f. = 0.69 (hexane:ethyl acetate 7:3). 1H-NMR (CDCl3) δ 8.19 (d, 1H, J = 4.8 Hz, HAr), 8.08 (d, 1H, J = 4.8 Hz, HAr), 7.76 (t, 1H, J = 4.7 Hz, HAr), 7.56 (d, 1H, J = 5.1 Hz, HAr), 7.44 (t, 1H, J = 4.5 Hz, HAr), 7.26 (t, 1H, J = 4.7 Hz, HAr), 6.78–6.68 (m, 2H, HAr), 6.47 (bs, 2H, NH2); 13C-NMR (CDCl3) δ 159.4, 158.0, 149.8, 146.7, 136.5, 133.7, 129.7, 128.7, 127.8, 126.4, 116.9, 116.8, 116.6, 110.1; IR (KBr): cm−1 3448, 3311, 1743, 1625, 1592, 1550, 1489, 1471, 1447, 1356, 1331, 1310, 1235, 1220, 1167, 1054, 1013; HRMS (ESI): m/z [M+H]+ calcd for C14H11N2O2: 239.08150, found: 239.08128;
3.1.11. The Synthesis of methyl 2-amino-5-chlorobenzoate (20)
To stirred slurry of 6-chloro-1H-benzo[d][1,3]oxazine-2,4-dione (13a, 591 mg, 3 mmol, 1 equiv.) in methanol (30 mL), sodium methoxide (486 mg, 9 mmol, 3 equiv.) was added, and the reaction mixture was refluxed for 3 h. After cooling down, the excess of solvent was evaporated under reduced pressure; the obtained residue was treated with water (100 mL) and extracted with ethyl acetate (3 × 50 mL). Combined organic layers were washed with water (1 × 100 mL), brine (1 × 50 mL) and dried over anhydrous magnesium sulfate. Evaporation of solvent gave pure product as slight yellow oil which solidified upon storage. Yield 97% (538 mg), m.p. 70–71 °C, R.f. = 0.80 (hexane:ethyl acetate 7:3). 1H-NMR (CDCl3) δ 7.81 (d, 1H, J = 2.4 Hz, HAr), 7.19 (dd, 1H, J = 2.4, 9.0 Hz, HAr), 6.59 (d, 1H, J = 9.0 Hz, HAr), 5.73 (bs, 2H, NH2), 3.86 (s, 3H, Me); 13C-NMR (CDCl3) δ 167.7, 149.1, 134.2, 130.5, 120.7, 118.1, 111.6, 51.9; HRMS (ESI): m/z [M+H]+ calcd for C8H9ClNO2: 186.03163, 188.02868 found: 186.03171, 188.02870.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
