Iron-Catalysed C(sp2)-H Borylation Enabled by Carboxylate Activation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
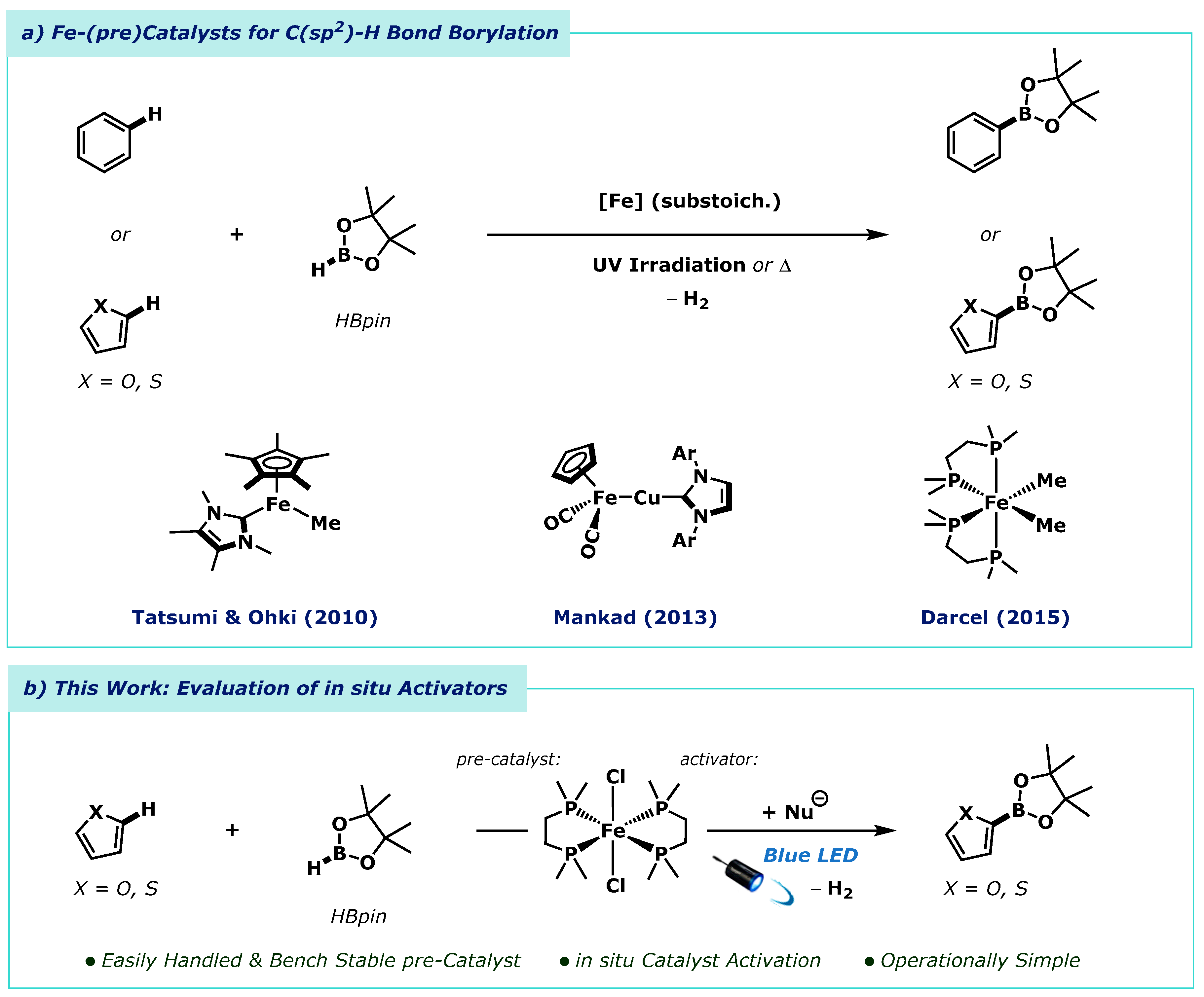
:1. Introduction
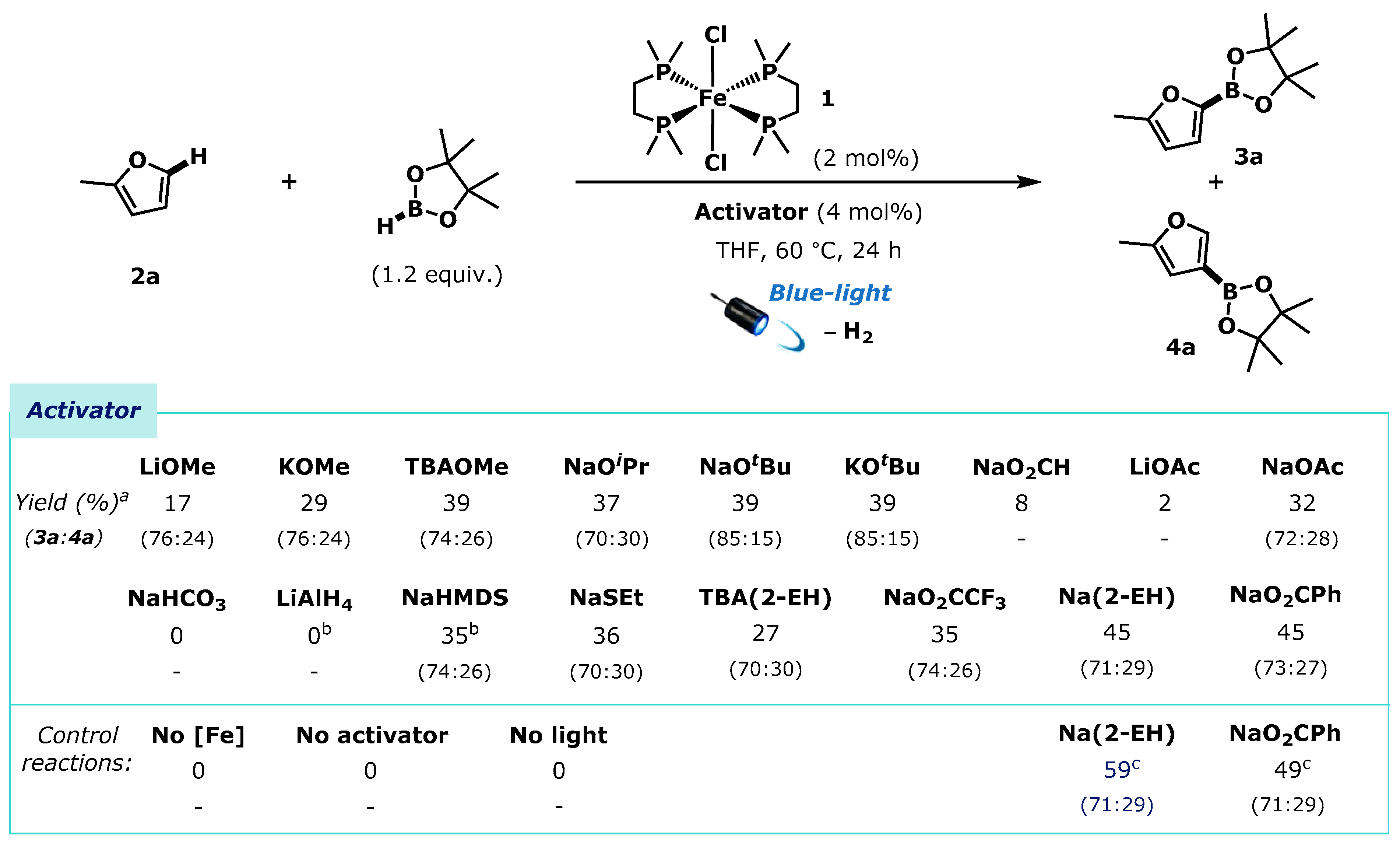
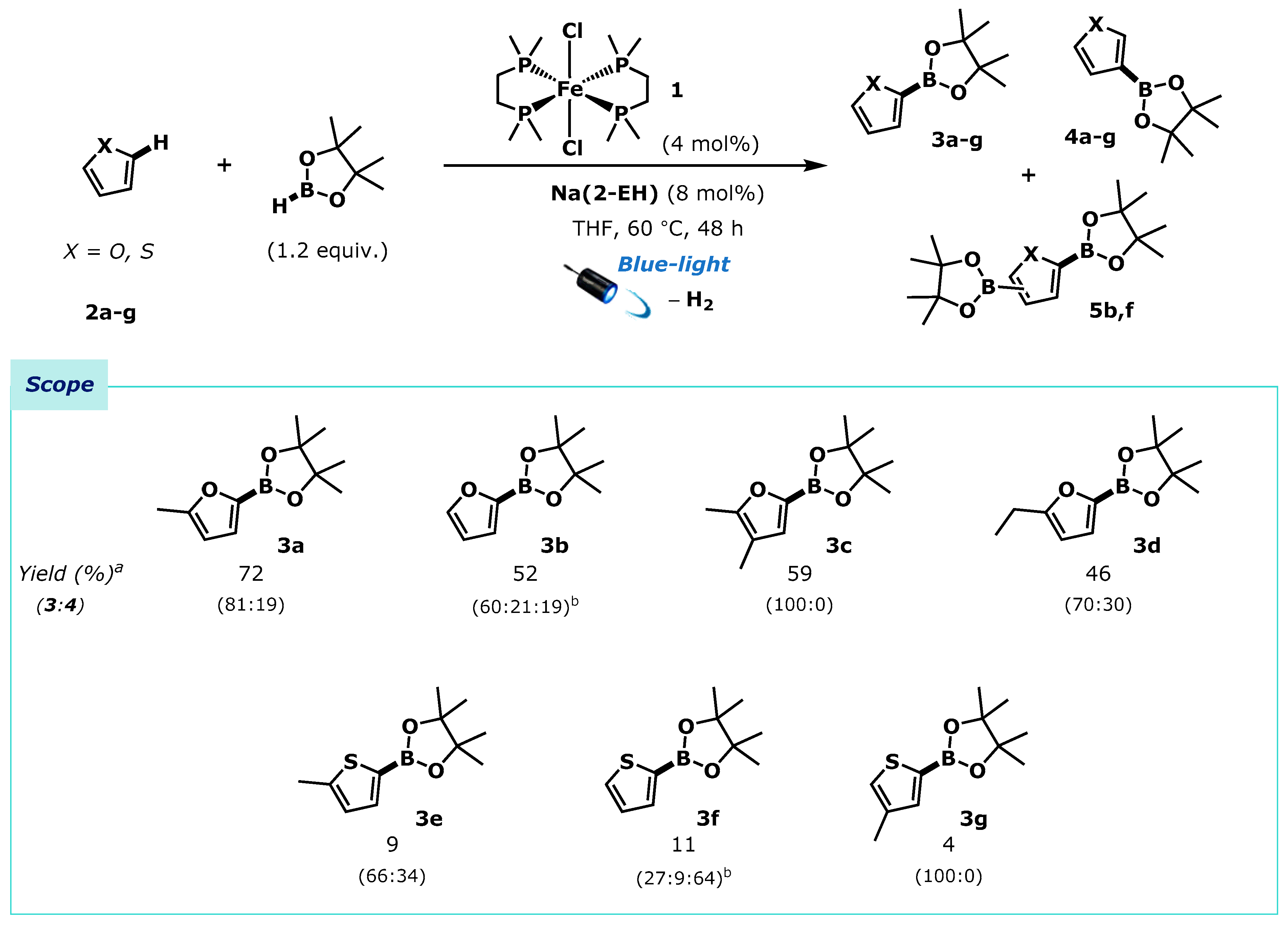
2. Results
2.1. Substrate Scope
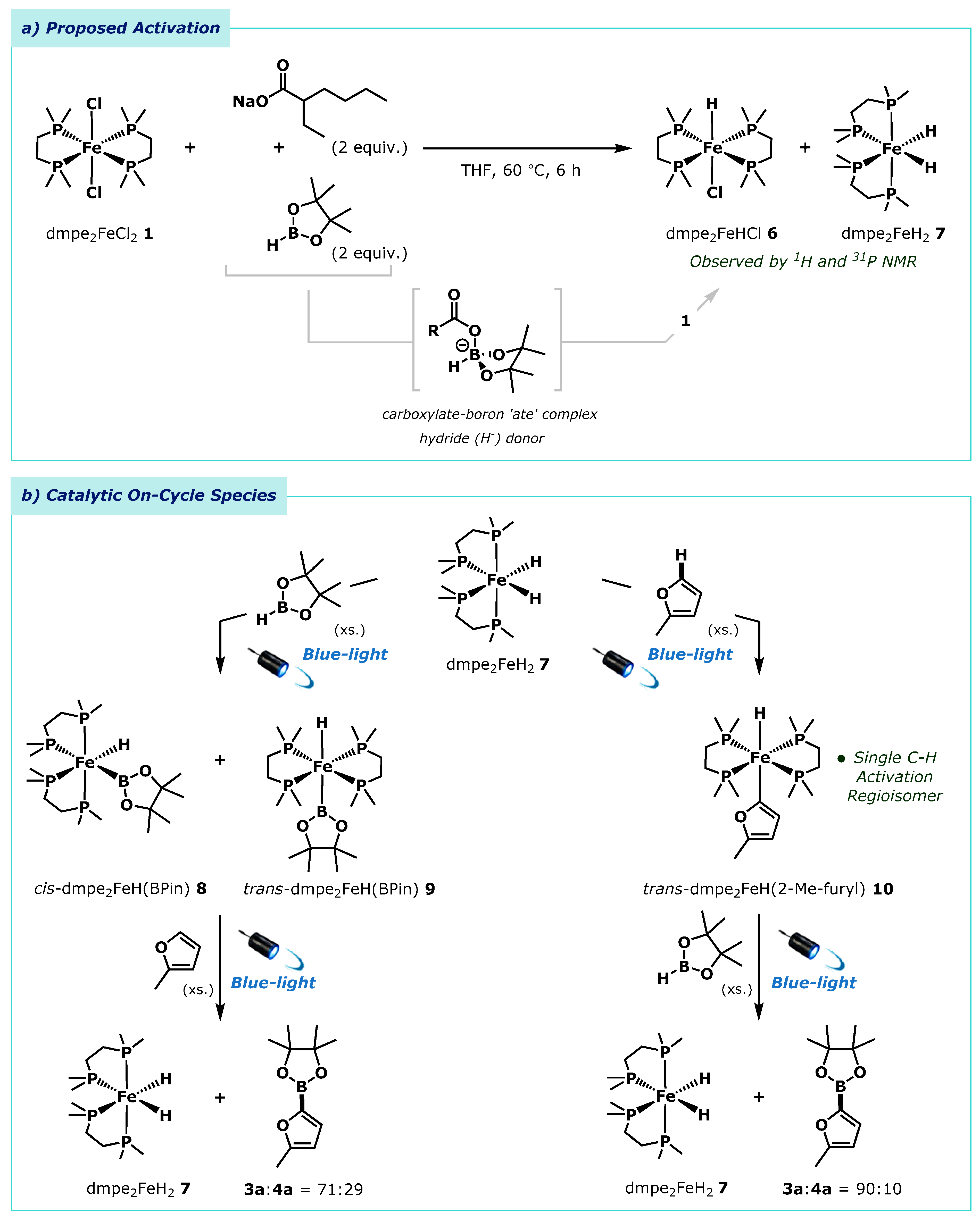
2.2. Mechanistic Investigations
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. Activator Synthesis
4.3. Pre-catalyst Synthesis
4.4. General Borylation Procedure
4.5. Characterisation of Borylated Products
4.5.1. 2-Methylfuran Derivatives
4.5.2. Furan Derivatives
4.5.3. 2.3-Dimethylfuran Derivatives
4.5.4. 2-Ethylfuran Derivatives
4.5.5. 2-Methylthiophene Derivatives
4.5.6. Thiophene Derivatives
4.5.7. 3-Methylthiophene Derivatives
4.6. Mechanistic Investigations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kakiuchi, F.; Chatani, N. Catalytic Methods for C-H Bond Functionalization: Application in Organic Synthesis. Adv. Synth. Catal. 2003, 345, 1077–1101. [Google Scholar] [CrossRef]
- Godula, K.; Sames, D. C-H bond functionalization in complex organic synthesis. Science 2006, 312, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Lyons, T.W.; Sanford, M.S. Palladium-catalyzed ligand-directed C-H functionalization reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, L.; O’Hara, F.; Gaunt, M.J. Recent developments in natural product synthesis using metal-catalysed C-H bond functionalisation. Chem. Soc. Rev. 2011, 40, 1885–1898. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Yamaguchi, A.D.; Itami, K. C-H bond functionalization: Emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem. Int. Ed. 2012, 51, 8960–9009. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Morton, D. Recent Advances in C-H Functionalization. J. Org. Chem. 2016, 81, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roudesly, F.; Oble, J.; Poli, G. Metal-catalyzed C-H activation/functionalization: The fundamentals. J. Mol. Catal. A Chem. 2017, 426, 275–296. [Google Scholar] [CrossRef]
- Su, B.; Cao, Z.C.; Shi, Z.J. Exploration of Earth-Abundant Transition Metals (Fe, Co, and Ni) as Catalysts in Unreactive Chemical Bond Activations. Acc Chem. Res. 2015, 48, 886–896. [Google Scholar] [CrossRef]
- Cera, G.; Ackermann, L. Iron-Catalyzed C–H Functionalization Processes. Top. Curr. Chem. 2016, 374, 57. [Google Scholar] [CrossRef]
- Shang, R.; Ilies, L.; Nakamura, E. Iron-Catalyzed C-H Bond Activation. Chem. Rev. 2017, 117, 9086–9139. [Google Scholar] [CrossRef]
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C-H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef] [PubMed]
- Loup, J.; Dhawa, U.; Pesciaioli, F.; Wencel-Delord, J.; Ackermann, L. Enantioselective C–H Activation with Earth-Abundant 3d Transition Metals. Angew. Chem. Int. Ed. 2019, 58, 12803–12818. [Google Scholar] [CrossRef] [PubMed]
- Mkhalid, I.A.I.; Barnard, J.H.; Marder, T.B.; Murphy, J.M.; Hartwig, J.F. C-H activation for the construction of C-B bonds. Chem. Rev. 2010, 110, 890–931. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, J.W.B.; Watson, A.J.B. Recent Developments in Organoboron Chemistry: Old Dogs, New Tricks. Chemistry 2017, 3, 31–55. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Wang, G.; Zhang, S.; Wang, H.; Wang, L.; Liu, L.; Jiao, J.; Li, P. Recent advances in catalytic C–H borylation reactions. Tetrahedron 2017, 73, 7123–7157. [Google Scholar] [CrossRef]
- Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N.R.; Hartwig, J.F. Mild iridium-catalyzed borylation of arenes. High turnover numbers, room temperature reactions, and isolation of a potential intermediate. J. Am. Chem. Soc. 2002, 124, 390–391. [Google Scholar] [CrossRef]
- Chotana, G.A.; Kallepalli, V.A.; Maleczka, R.E.; Smith, M.R. Iridium-catalyzed borylation of thiophenes: Versatile, synthetic elaboration founded on selective C-H functionalization. Tetrahedron 2008, 64, 6103–6114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwig, J.F. Regioselectivity of the borylation of alkanes and arenes. Chem. Soc. Rev. 2011, 40, 1992–2002. [Google Scholar] [CrossRef]
- Hartwig, J.F. Borylation and silylation of C-H bonds: A platform for diverse C-H bond functionalizations. Acc. Chem. Res. 2012, 45, 864–873. [Google Scholar] [CrossRef]
- Preshlock, S.M.; Plattner, D.L.; Maligres, P.E.; Krska, S.W.; Maleczka, R.E.; Smith, M.R. A traceless directing group for C-H borylation. Angew. Chem. Int. Ed. 2013, 52, 12915–12919. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.A.; Hartwig, J.F. Iridium-catalyzed C-H borylation of heteroarenes: Scope, regioselectivity, application to late-stage functionalization, and mechanism. J. Am. Chem. Soc. 2014, 136, 4287–4299. [Google Scholar] [CrossRef] [PubMed]
- Sadler, S.A.; Tajuddin, H.; Mkhalid, I.A.I.; Batsanov, A.S.; Albesa-Jove, D.; Cheung, M.S.; Maxwell, A.C.; Shukla, L.; Roberts, B.; Blakemore, D.C.; et al. Iridium-catalyzed C-H borylation of pyridines. Org. Biomol. Chem. 2014, 12, 7318–7327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Segawa, Y.; Itami, K. Para C-H borylation of benzene derivatives by a bulky iridium catalyst. J. Am. Chem. Soc. 2015, 137, 5193–5198. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Semba, K.; Nakao, Y. Para-Selective C–H Borylation of (Hetero)Arenes by Cooperative Iridium/Aluminum Catalysis. Angew. Chem. Int. Ed. 2017, 56, 4853–4857. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Jiang, Y.; Kuang, C.; Wang, S.; Liu, H.; Zhang, Y.; Wang, J. Nano-Fe2O3-catalyzed direct borylation of arenes. Chem. Commun. 2010, 46, 3170–3172. [Google Scholar] [CrossRef]
- Zhang, H.; Hagihara, S.; Itami, K. Aromatic C-H borylation by nickel catalysis. Chem. Lett. 2015, 44, 779–781. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, T.; Tobisu, M.; Chatani, N. Nickel-catalyzed borylation of arenes and indoles via C-H bond cleavage. Chem. Commun. 2015, 51, 6508–6511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzacano, T.J.; Mankad, N.P. Thermal C-H borylation using a CO-free iron boryl complex. Chem. Commun. 2015, 51, 5379–5382. [Google Scholar] [CrossRef]
- Obligacion, J.V.; Semproni, S.P.; Pappas, I.; Chirik, P.J. Cobalt-Catalyzed C(sp2)-H Borylation: Mechanistic Insights Inspire Catalyst Design. J. Am. Chem. Soc. 2016, 138, 10645–10653. [Google Scholar] [CrossRef]
- Léonard, N.G.; Bezdek, M.J.; Chirik, P.J. Cobalt-Catalyzed C(sp2)-H borylation with an air-stable, readily prepared terpyridine cobalt(II) Bis(acetate) precatalyst. Organometallics 2017, 36, 142–150. [Google Scholar] [CrossRef]
- Yoshigoe, Y.; Kuninobu, Y. Iron-Catalyzed ortho-Selective C-H Borylation of 2-Phenylpyridines and Their Analogs. Org. Lett. 2017, 19, 3450–3453. [Google Scholar] [CrossRef] [PubMed]
- Obligacion, J.V.; Bezdek, M.J.; Chirik, P.J. C(sp2)-H Borylation of Fluorinated Arenes Using an Air-Stable Cobalt Precatalyst: Electronically Enhanced Site Selectivity Enables Synthetic Opportunities. J. Am. Chem. Soc. 2017, 139, 2825–2832. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, M.; Kusaka, H.; Yuge, H. Iron-catalyzed versatile and efficient C(sp2)-H borylation. Chem. Lett. 2019, 48, 898–901. [Google Scholar] [CrossRef] [Green Version]
- Agahi, R.; Challinor, A.J.; Dunne, J.; Docherty, J.H.; Carter, N.B.; Thomas, S.P. Regiodivergent hydrosilylation, hydrogenation, [2π + 2π]-cycloaddition and C-H borylation using counterion activated earth-abundant metal catalysis. Chem. Sci. 2019, 10, 5079–5084. [Google Scholar] [CrossRef] [Green Version]
- Hatanaka, T.; Ohki, Y.; Tatsumi, K. C-H bond activation/borylation of furans and thiophenes catalyzed by a half-sandwich iron N-heterocyclic carbene complex. Chemistry 2010, 5, 1657–1666. [Google Scholar] [CrossRef]
- Mazzacano, T.J.; Mankad, N.P. Base metal catalysts for photochemical C-H borylation that utilize metal-metal cooperativity. J. Am. Chem. Soc. 2013, 135, 17258–17261. [Google Scholar] [CrossRef]
- Dombray, T.; Werncke, C.G.; Jiang, S.; Grellier, M.; Vendier, L.; Bontemps, S.; Sortais, J.B.; Sabo-Etienne, S.; Darcel, C. Iron-catalyzed C-H borylation of arenes. J. Am. Chem. Soc. 2015, 137, 4062–4065. [Google Scholar] [CrossRef]
- Allen, O.R.; Dalgarno, S.J.; Field, L.D.; Jensen, P.; Turnbull, A.J.; Willis, A.C. Addition of CO2 to alkyl iron complexes, Fe(PP)2Me2. Organometallics 2008, 27, 2092–2098. [Google Scholar] [CrossRef]
- Docherty, J.H.; Peng, J.; Dominey, A.P.; Thomas, S.P. Activation and discovery of earth-abundant metal catalysts using sodium tert-butoxide. Nat. Chem. 2017, 9, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Buys, I.E.; Field, L.D.; Hambley, T.W.; McQueen, A.E.D.J. Photochemical reactions of [cis-Fe(H)2(Me2PCH2CH2PMe2)2] with thiophenes: Insertion into C–H and C–S bonds. Chem. Soc. Chem. Commun. 1994, 5, 557–558. [Google Scholar] [CrossRef]
- Query, I.P.; Squier, P.A.; Larson, E.M.; Isley, N.A.; Clark, T.B. Alkoxide-catalyzed reduction of ketones with pinacolborane. J. Org. Chem. 2011, 76, 6452–6456. [Google Scholar] [CrossRef]
- Elton, T.E.; Ball, G.E.; Bhadbhade, M.; Field, L.D.; Colbran, S.B. Evaluation of Organic Hydride Donors as Reagents for the Reduction of Carbon Dioxide and Metal-Bound Formates. Organometallics 2018, 37, 3972–3982. [Google Scholar] [CrossRef]
- Wollenburg, M.; Moock, D.; Glorius, F. Hydrogenation of Borylated Arenes. Angew. Chem. Int. Ed. 2019, 58, 6549–6553. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiao, L. Pyridine-Catalyzed Radical Borylation of Aryl Halides. J. Am. Chem. Soc. 2017, 139, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Miyaura, N.; Ishiyama, T. Process for the Production of Hetereoaryl-type Boron Compounds with Iridium Catalyst. European Patent EP1481978A1, 1 December 2004. [Google Scholar]
- Xue, C.; Luo, Y.; Teng, H.; Ma, Y.; Nishiura, M.; Hou, Z. Ortho-Selective C–H Borylation of Aromatic Ethers with Pinacol-borane by Organo Rare-Earth Catalysts. ACS Catalysis. 2018, 8, 5017–5022. [Google Scholar] [CrossRef]
- Kato, T.; Kuriyama, S.; Nakajima, K.; Nishibayashi, Y. Catalytic C–H Borylation Using Iron Complexes Bearing 4,5,6,7-Tetrahydroisoindol-2-ide-Based PNP-Type Pincer Ligand. Chem Asian J. 2019, 14, 2097–2101. [Google Scholar] [CrossRef]
- Tobisu, M.; Igarashi, T.; Chatani, N. Iridium/N-heterocyclic carbene-catalyzed C–H borylation of arenes by diisopropylaminoborane. Beilstein J. Org. Chem. 2016, 12, 654–661. [Google Scholar] [CrossRef] [Green Version]
- Ratniyom, J.; Dechnarong, N.; Yotphan, S.; Kiatisevi, S. Convenient Synthesis of Arylboronates through a Synergistic Pd/Cu-Catalyzed Miyaura Borylation Reaction under Atmospheric Conditions. Eur. J. Org. Chem. 2014, 7, 1381–1385. [Google Scholar] [CrossRef]
Sample Availability: Samples of the pre-catalyst 1 are available from the authors. |




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Britton, L.; Docherty, J.H.; Dominey, A.P.; Thomas, S.P. Iron-Catalysed C(sp2)-H Borylation Enabled by Carboxylate Activation. Molecules 2020, 25, 905. https://doi.org/10.3390/molecules25040905
Britton L, Docherty JH, Dominey AP, Thomas SP. Iron-Catalysed C(sp2)-H Borylation Enabled by Carboxylate Activation. Molecules. 2020; 25(4):905. https://doi.org/10.3390/molecules25040905
Chicago/Turabian StyleBritton, Luke, Jamie H. Docherty, Andrew P. Dominey, and Stephen P. Thomas. 2020. "Iron-Catalysed C(sp2)-H Borylation Enabled by Carboxylate Activation" Molecules 25, no. 4: 905. https://doi.org/10.3390/molecules25040905
APA StyleBritton, L., Docherty, J. H., Dominey, A. P., & Thomas, S. P. (2020). Iron-Catalysed C(sp2)-H Borylation Enabled by Carboxylate Activation. Molecules, 25(4), 905. https://doi.org/10.3390/molecules25040905