
C-H Functionalization via Iron-Catalyzed Carbene-Transfer Reactions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
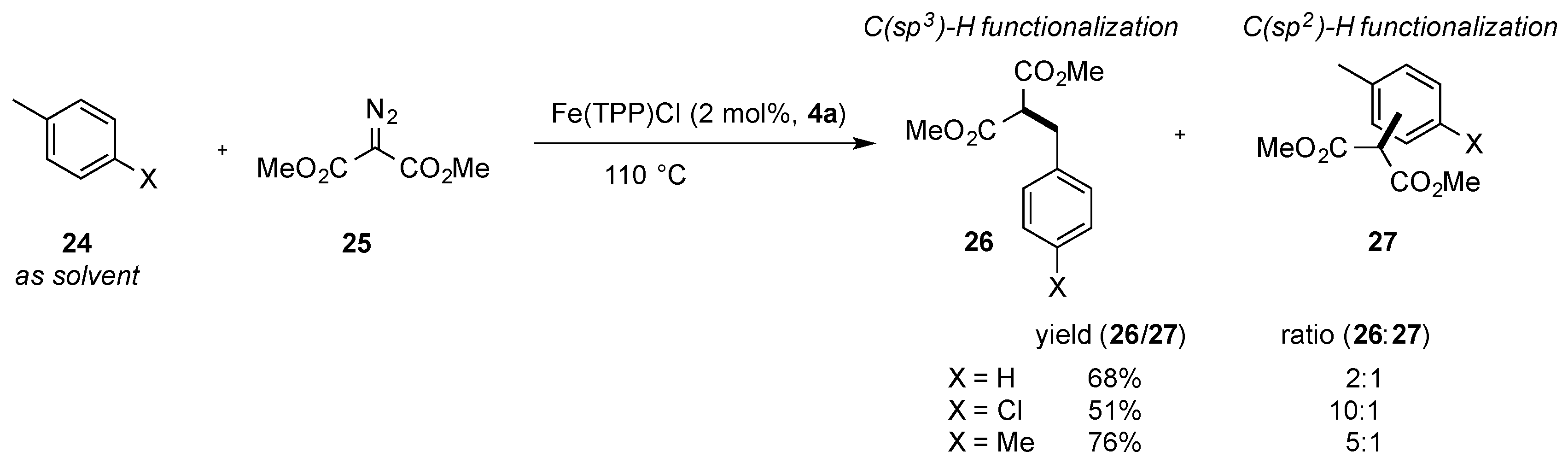{kind=link}
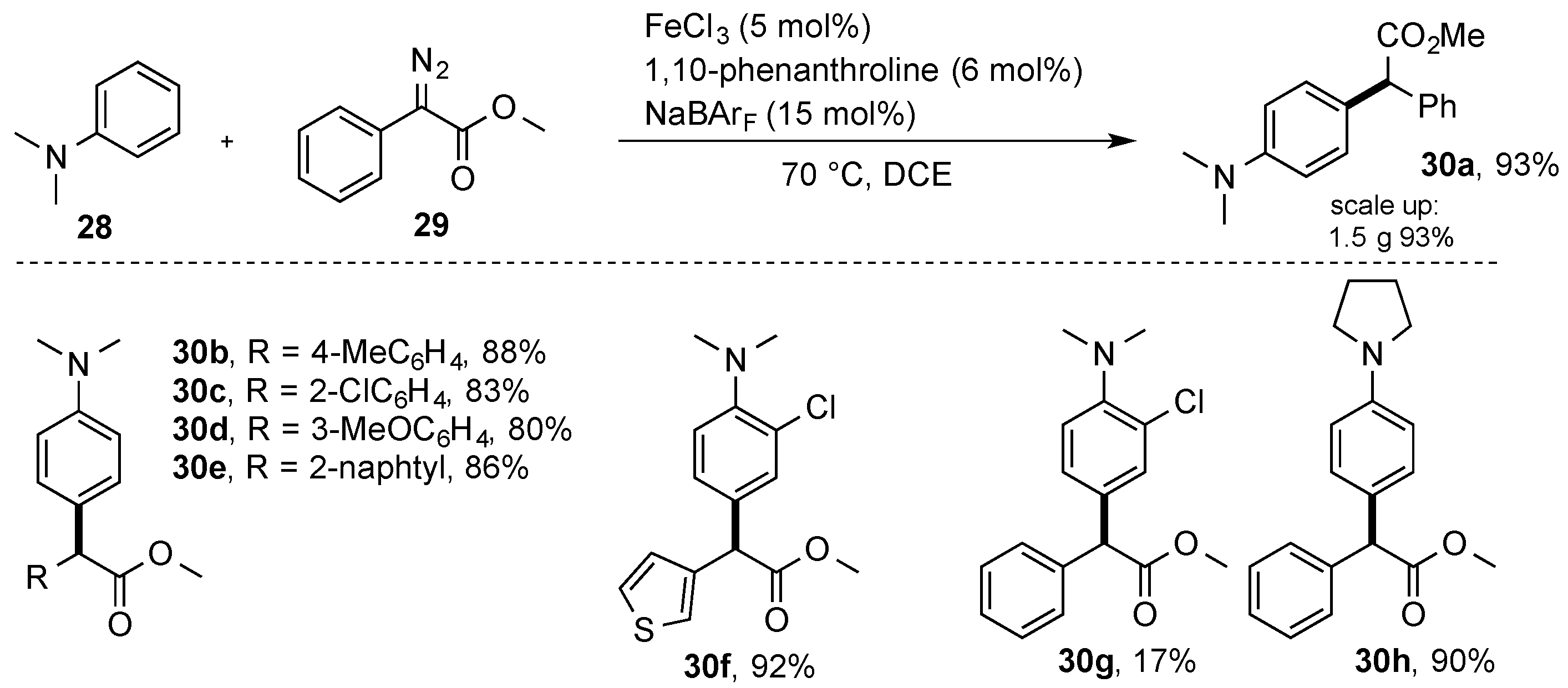{kind=link}
{kind=link}
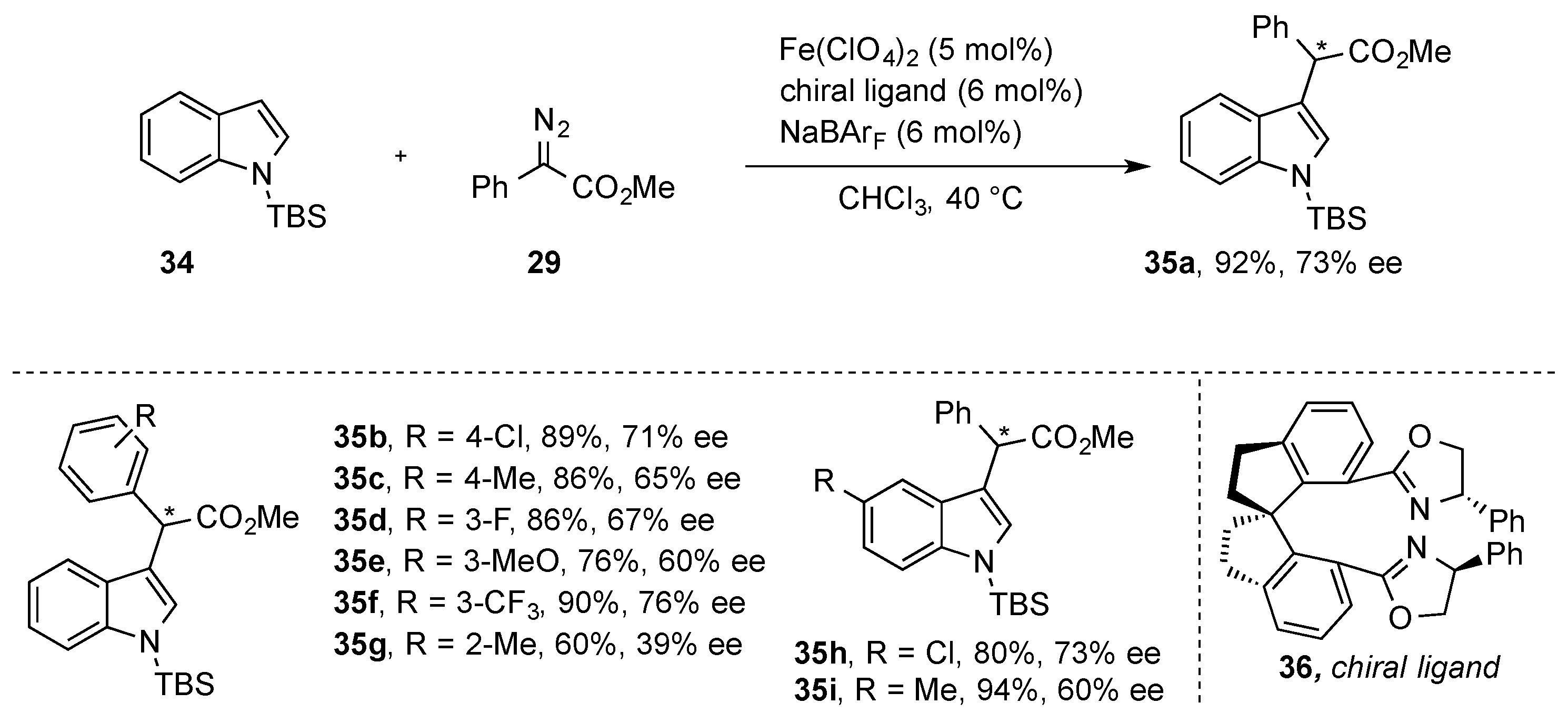{kind=link}
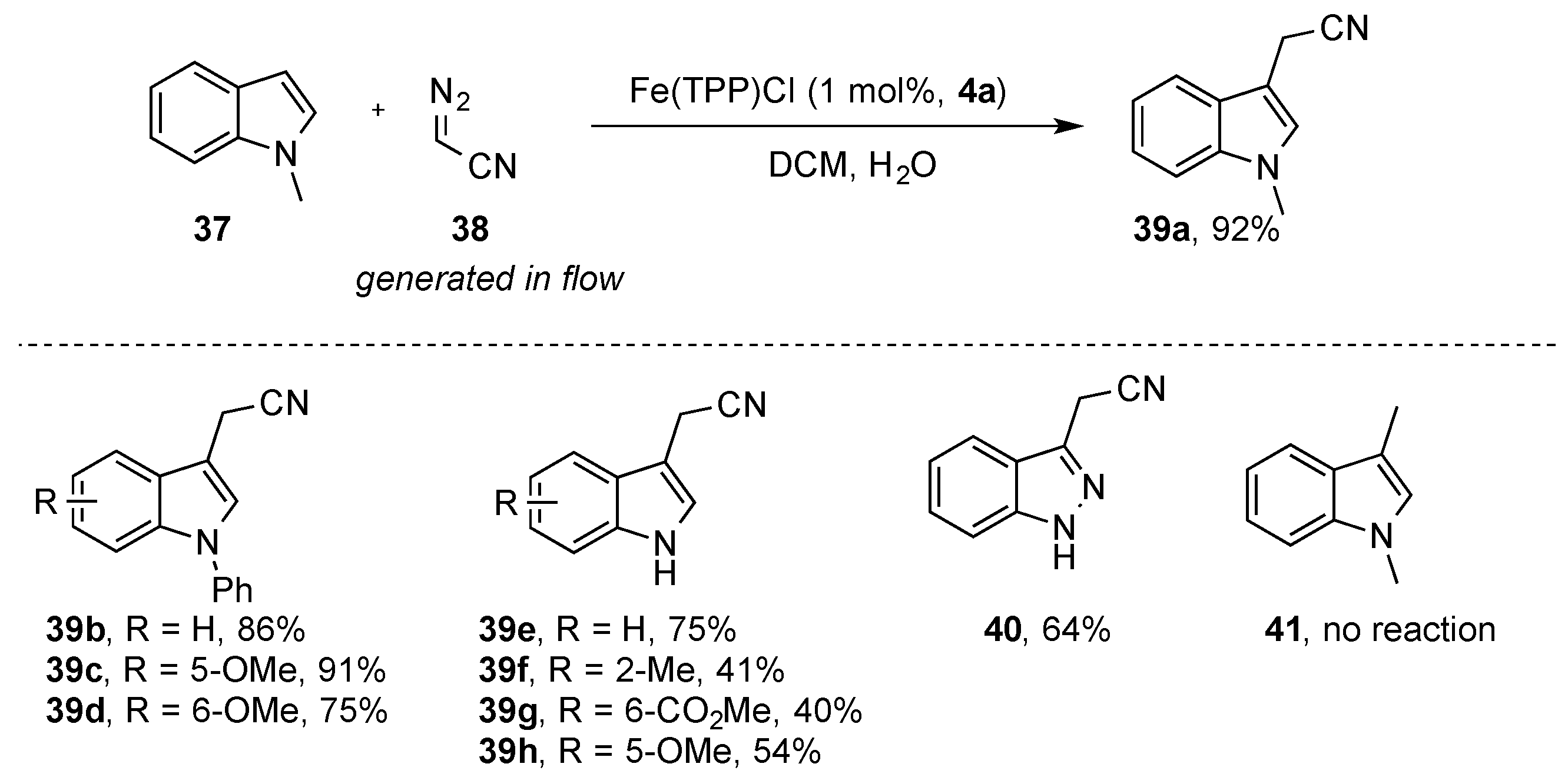{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
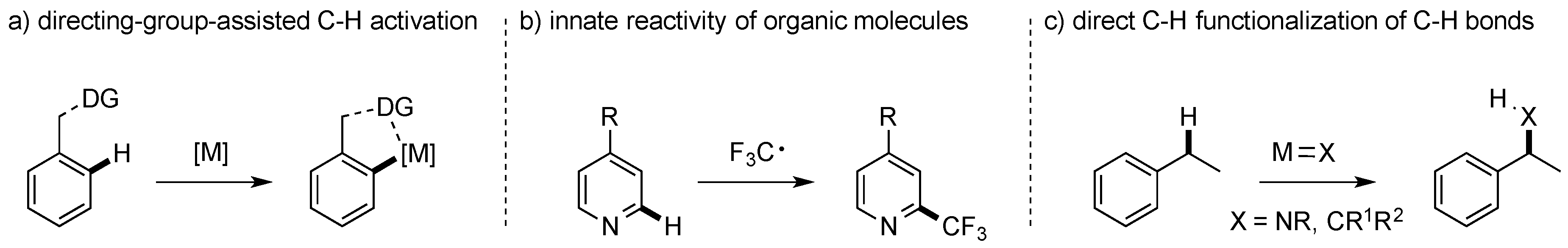
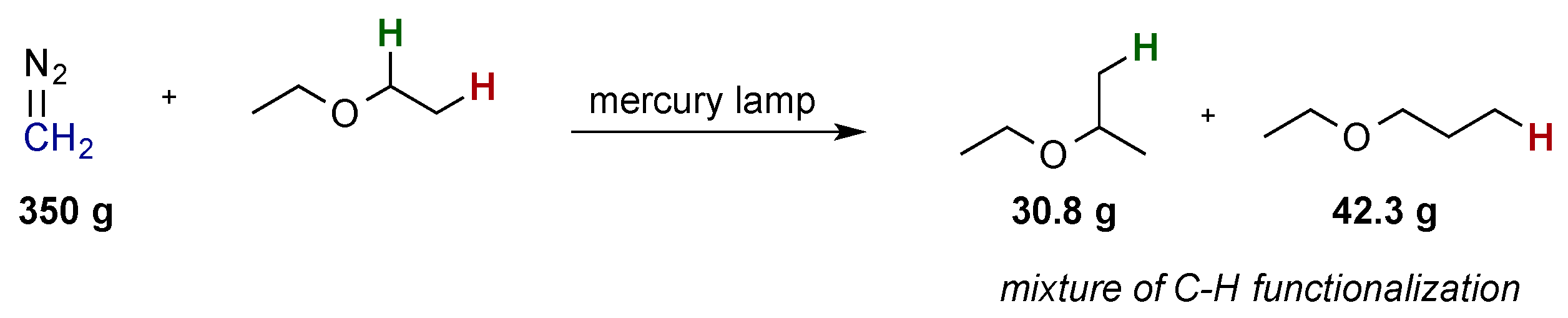
:1. Introduction
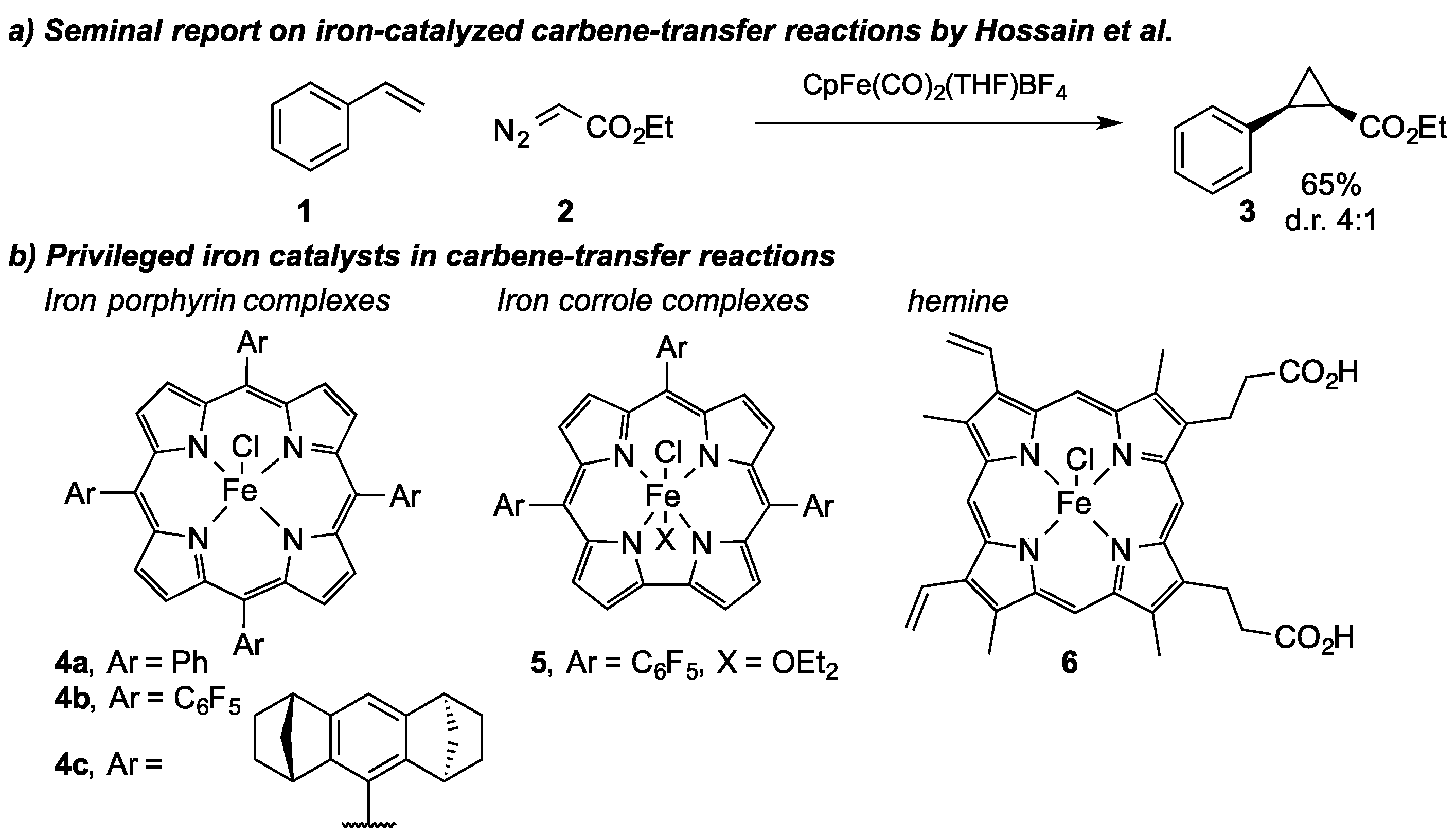
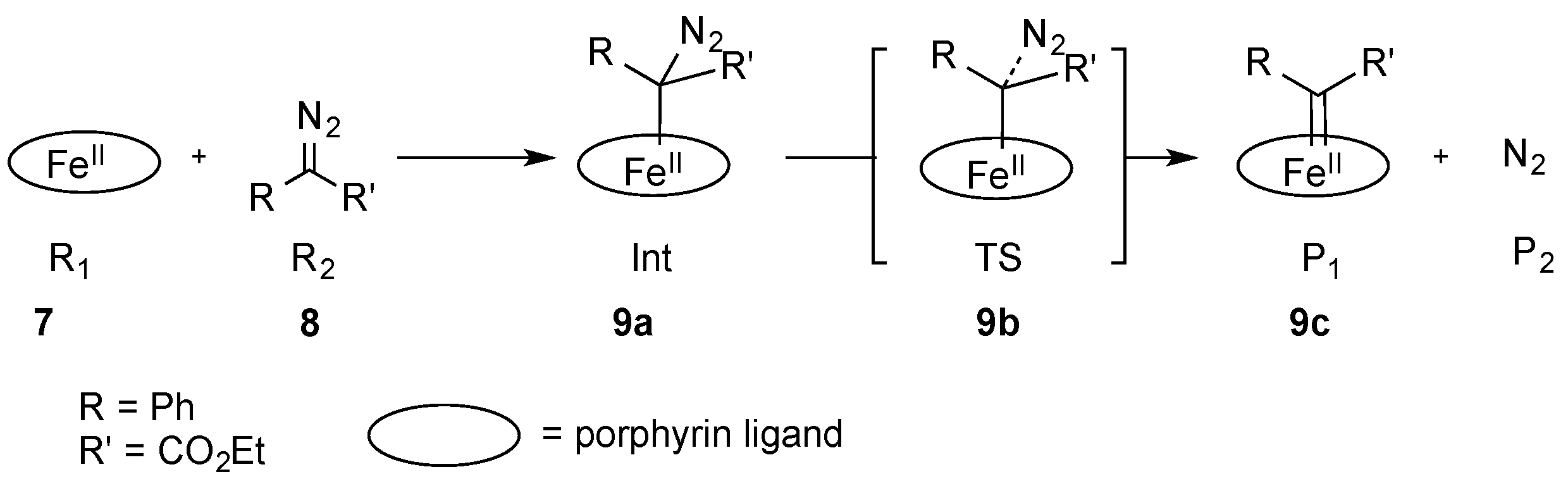
2. Iron in Carbene-Transfer Reactions
3. Stoichiometric C-H Functionalization
4. C-H Functionalization Reactions of Aromatic C-H Bonds
5. C-H Functionalization Reactions of Heteroaromatic C-H Bonds
6. C-H Functionalization Reactions of Aliphatic C-H Bonds
7. Biocatalytic C-H Functionalization Reactions of Aromatic C-H Bonds
8. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Norinder, J.; Matsumoto, A.; Yishikai, N.; Nakamura, E. Iron-Catalyzed Direct Arylation trough Directed C-H Bond Activation. J. Am. Chem. Soc. 2008, 130, 5858–5859. [Google Scholar] [CrossRef] [PubMed]
- Wencel-Delord, J.; Dröger, T.; Liu, F.; Glorius, F. Towads mild metal-catalyzed C-H activation. Chem. Soc. Rev. 2011, 40, 4740–4761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, Y.; Jie, X.; Zhao, H.; Lim, G.; Su, W. Recent advantages in directed C-H functionalizations using monodentate nitrogen-based directing groups. Org. Chem. Front. 2014, 1, 843–895. [Google Scholar] [CrossRef]
- Jia, C.; Kitamura, T.; Fujiwara, Y. Catalytic Functionalization of Arenes and Alkanes via C-H Bond Activation. Acc. Chem. Res. 2001, 34, 633–639. [Google Scholar] [CrossRef]
- Guptill, D.M.; Davies, H.M.L. 2,2,2-Trichloroethyl Aryldiazoacetates as Robust Reagents for the Enantioselective C-H Functionalization of Methyl Ethers. J. Am. Chem. Soc. 2014, 136, 17718–17721. [Google Scholar] [CrossRef]
- Pan, S.; Shibata, T. Recent Advances in Iridum-Catalyzed Alkylation of C-H and N-H Bonds. Acs Catal. 2013, 3, 704–712. [Google Scholar] [CrossRef]
- Ammann, S.E.; Rice, G.T.; White, M.C. Terminal Olefins to Chromans, Isochromans and Pyrans via Allylic C-H Oxidation. J. Am. Chem. Soc. 2014, 136, 10834–10837. [Google Scholar] [CrossRef] [Green Version]
- Burbidge, E.; Burbidge, G.R.; Fowler, A.W.; Hoyle, F. Synthesis of the Elements in stars. Rev. Mod. Phys. 1957, 29, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Tansition Metals for C-H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef]
- Shang, R.; Illies, L.; Nakamura, E. Iron-Catalyzed C-H Bond Activation. Chem. Rev. 2017, 117, 9086–9139. [Google Scholar] [CrossRef]
- Zhu, S.-F.; Zhou, Q.-L. Iron-catalyzed transformations of diazo compounds. Natl. Sci. Rev. 2014, 1, 580–603. [Google Scholar] [CrossRef] [Green Version]
- Bauer, I.; Knölker, H.-J. Iron Catalysis in Organic Synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar] [CrossRef] [PubMed]
- Plietker, B. Iron Catalysis in Organic Chemistry: Reactions and Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Felg, A.L.; Lippard, S.J. Reactions of Non-Heme Iron(II) Centers with Dioxygen in Biology and Chemistry. Chem. Rev. 1994, 94, 759–805. [Google Scholar]
- Meerwein, H.; Rathjen, H.; Werner, H. Die Methylierung von RH-Verbindungen mittels Diazomethan unter Mitwirkung des Lichtes. Ber. Dtsch. Chem. Ges. 1942, 24, 136–137. [Google Scholar] [CrossRef]
- von E. Doering, W.; Knox, L.; Jones, M. Notes. Reaction of Methylene with Diethyl Ether and Tetrahydrofuran. J. Org. Chem. 1959, 24, 136–137. [Google Scholar] [CrossRef]
- Ford, A.; Miel, M.; Ring, A.; Slattery, C.N.; Maguire, A.R.; McKervey, M.A. Modern Organic Synthesis with α-Doazocarbonyl Compounds. Chem. Rev. 2015, 115, 9981–10080. [Google Scholar] [CrossRef]
- Doyle, M.P.; Duffy, R.; Ratnikov, M.; Zhou, L. Catalytic Carbene Insertion into C-H Bonds. Chem. Rev. 2010, 110, 704–724. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Manning, J.R. Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Morton, D. Guiding principles for site selective and stereoselective intramolecular C-H functionalization by donor/acceptor rhodium carbenes. Chem. Soc. Rev. 2011, 40, 1857–1869. [Google Scholar] [CrossRef]
- Empel, C.; Koenigs, R.M. Sustainable Carbene Transfer Reactions with Iron and Light. Synlett 2019, 30, 1929–1934. [Google Scholar] [CrossRef]
- Seitz, W.J.; Saha, A.K.; Hossain, M.M. Iron Lewis acid catalyzed cyclopropanation reaction of ethyl diazoacetate and olefins. Organometallics 1993, 12, 2604–2608. [Google Scholar] [CrossRef]
- Wolf, J.R.; Hamaker, C.G.; Djukic, J.-P.; Kodadek, T.; Woo, L.K. Shape and stereoselective cyclopropanation of alkenes catalyzed by iron porphyrins. J. Am. Chem. Soc. 1995, 117, 9194–9199. [Google Scholar] [CrossRef] [Green Version]
- Lai, T.-S.; Chan, F.-Y.; So, P.-K.; Ma, D.-L.; Wong, K.Y.; Che, C.-M. Alkene cyclopropanation catalyzed by Halterman iron porphyrin: Participation of organic based axial ligands. Dalton Trans. 2006, 40, 4845–4851. [Google Scholar] [CrossRef] [PubMed]
- Aviv, I.; Gross, Z. Corrole-based applications. Chem. Commun. 2007, 20, 1987–1999. [Google Scholar] [CrossRef] [PubMed]
- Morandi, B.; Carreira, E.M. Iron-Catalyzed Cyclopropanation with Trifluoroethylamine Hydrochloride and Olefines in Aqueous Media: In Situ Generation of Trifluoromethyl Diazomethane. Angew. Chem. Int. Ed. 2010, 49, 938–941. [Google Scholar] [CrossRef]
- Khade, R.L.; Zhang, Y. C-H Insertions by Iron Porphyrin Carbene: Basis Mechanism and Origin of Substrate Selectivity. Chem. Eur. J. 2017, 23, 17654–17658. [Google Scholar] [CrossRef]
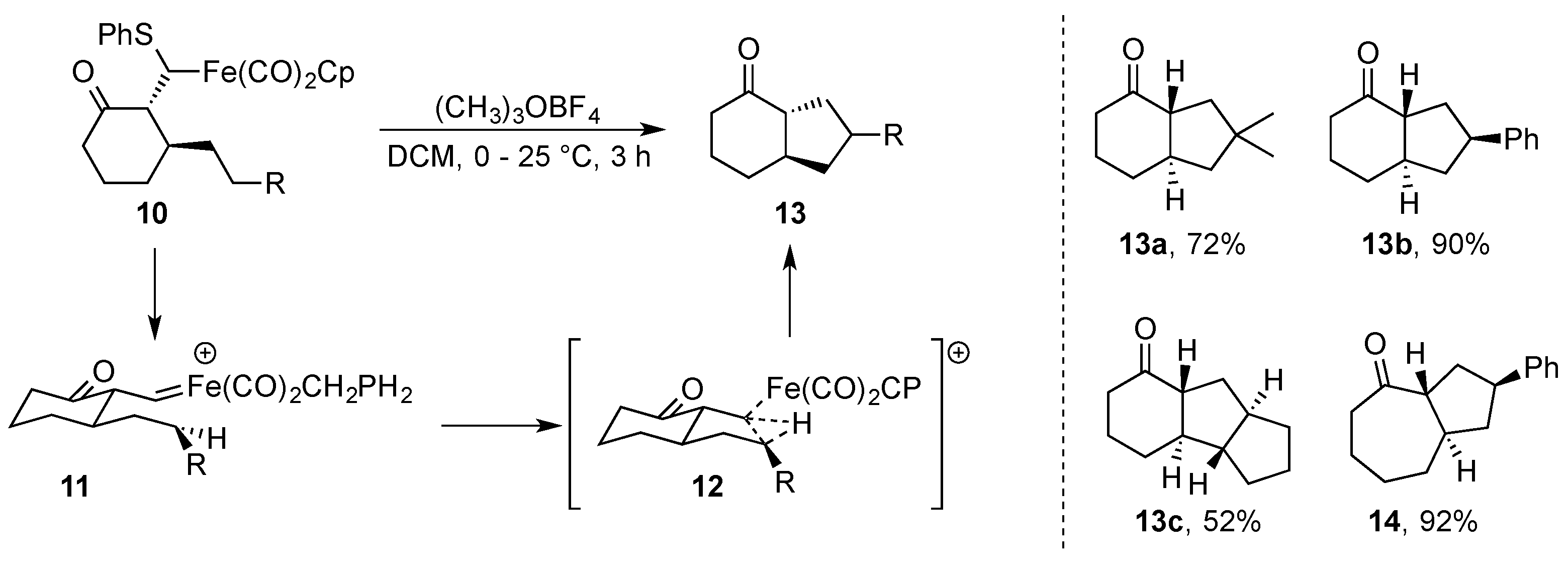
- Ishii, S.; Helquist, P. Intramolecular C-H Insertion Reaction of Iron Carbene Complexes as a General Method for Synthesis of Bicyclo[n.3.0]alkanones. Synlett 1997, 4, 508–510. [Google Scholar] [CrossRef]
- Ishii, S.; Zhao, S.; Nehta, G.; Knors, C.J.; Helquist, P. Intramolecular C-H insertion Reactions of (η5-Cyclopentadienyl)dicabonyliron Carbene Complexes: Scope of the Reaction and Application to the Synthesos of (±)-Sterpurene and (±)-Pentalene. J. Org. Chem. 2001, 66, 3449–3458. [Google Scholar] [CrossRef]
- Ishii, S.; Zhao, S.; Helquist, P. Stereochemical Probes of Intramolecular C-H Insertion Reaction of Iron-Carbene Complexes. J. Am. Chem. Soc. 2000, 122, 5897–5898. [Google Scholar] [CrossRef]
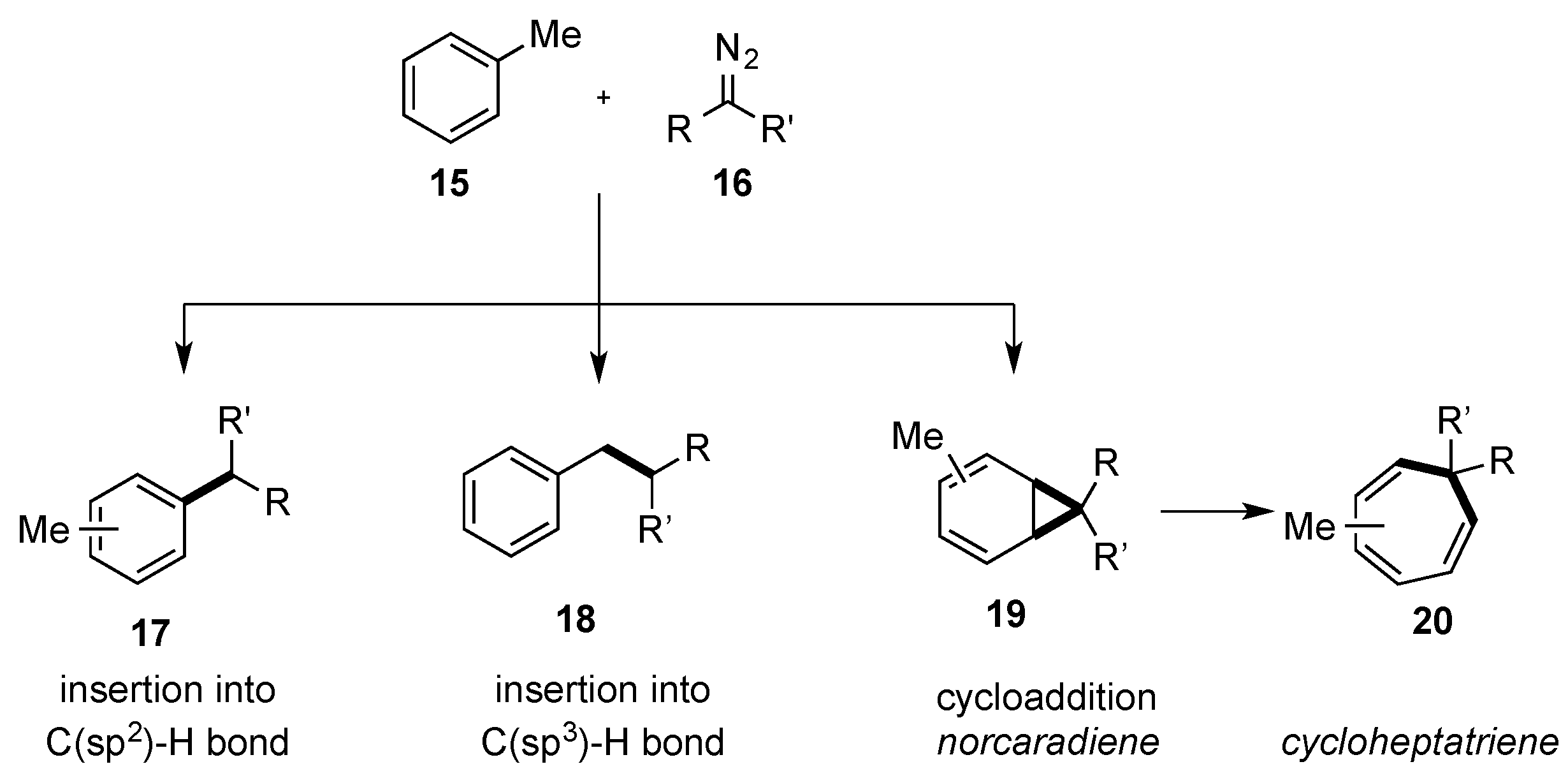
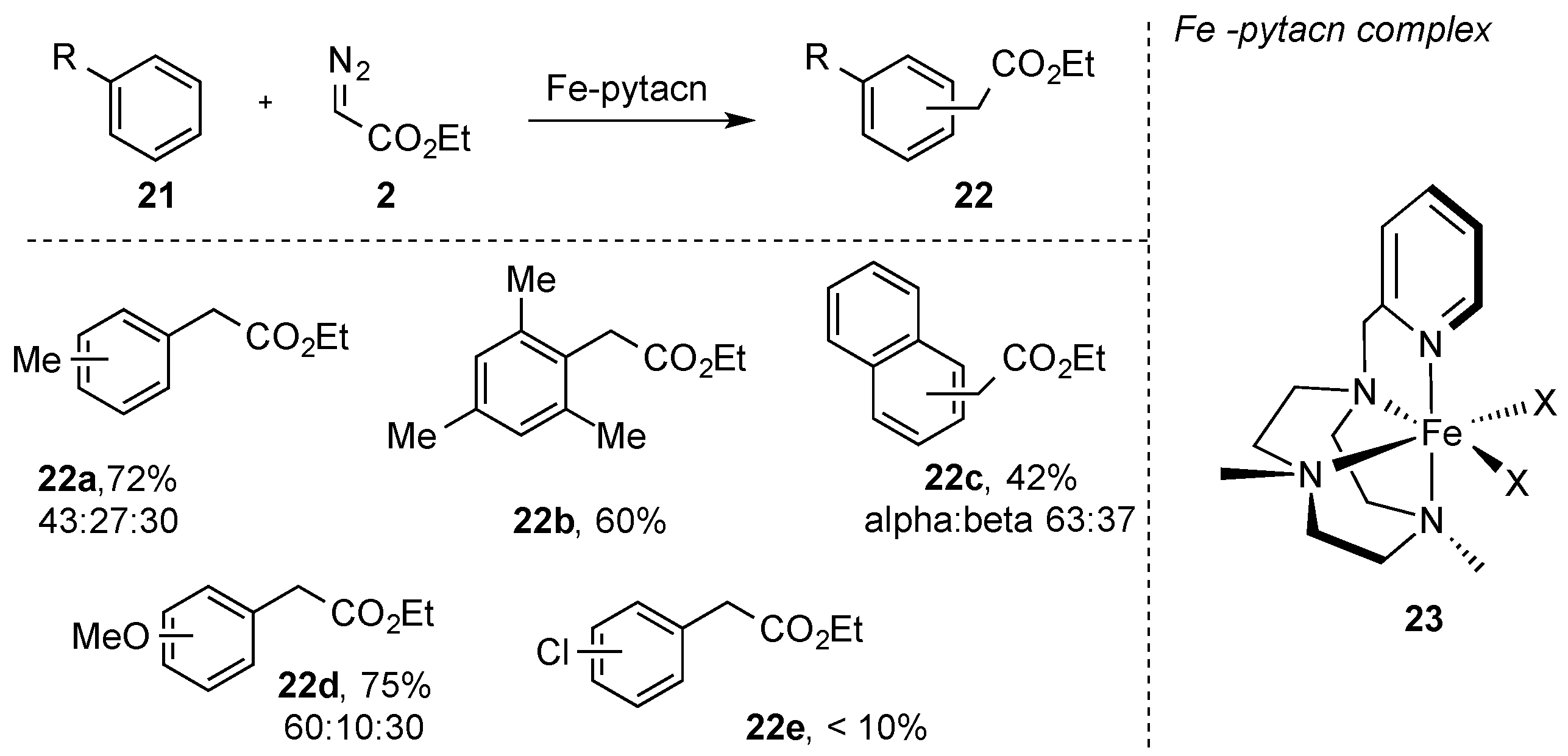
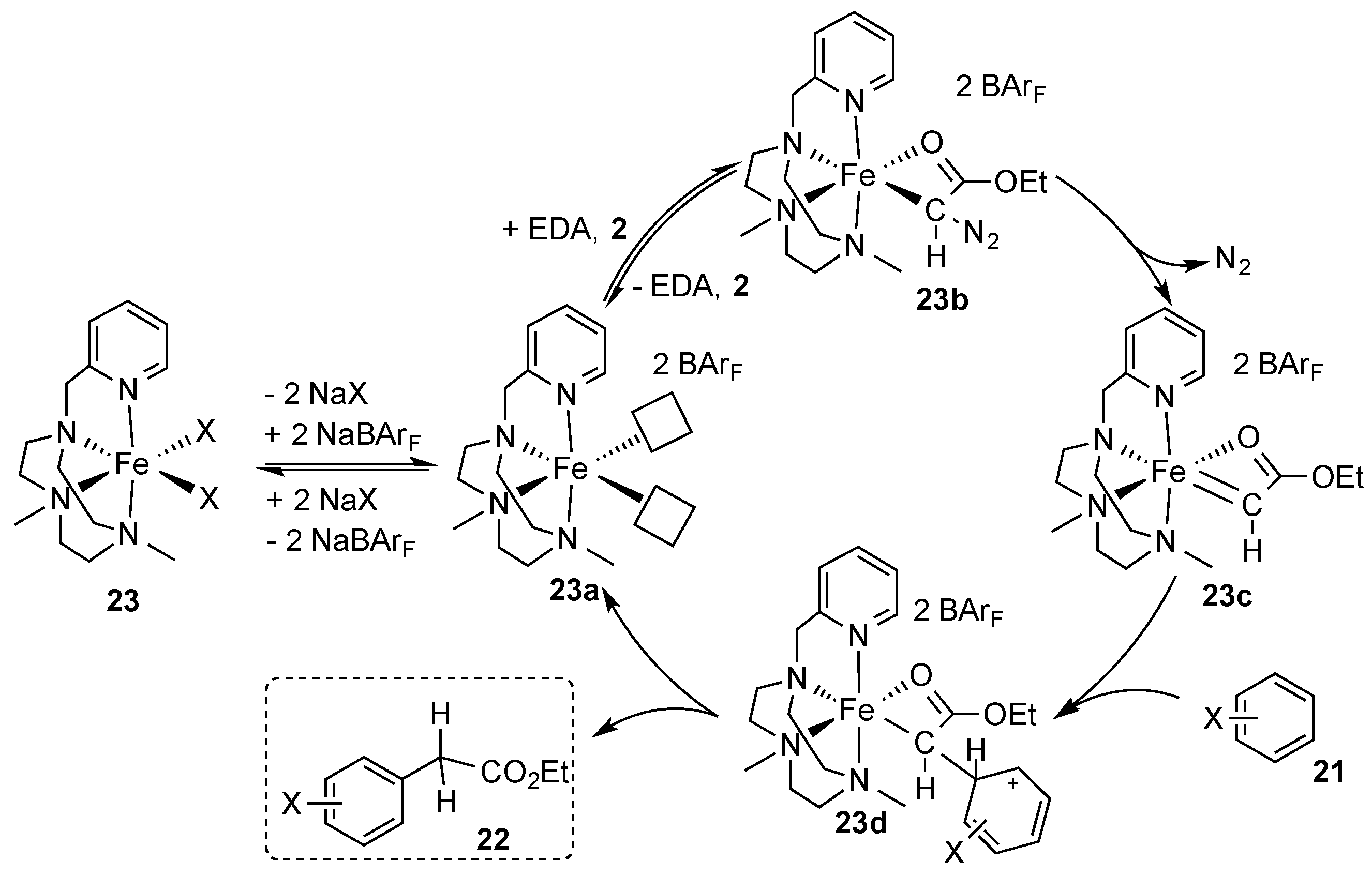
- Postils, V.; Rodriguez, M.; Sabenya, G.; Conde, A.; Mar Diaz-Requejo, M.; Pérez, P.J.; Costas, M.; Solà, M.; Luis, J.M. Mechanism of the Selective Fe-Catalyzed Arene Carbon-Hydrogen Bond Functionalization. Acs Catal. 2018, 8, 4313–4322. [Google Scholar] [CrossRef] [Green Version]
- Conde, A.; Sabenya, G.; Rodriguez, M.; Postil, V.; Luis, J.M.; Mar Diaz-Requejo, M.; Costas, M.; Pérez, P.J. Iron and Manganese Catalysts for the Selective Functionalization of Arene C(sp2)-H Bonds by Carbene Insertion. Angew. Chem Int. Ed. 2016, 55, 6530–6534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padwa, A.; Austin, D.J.; Price, A.T.; Semones, M.A.; Doyle, M.P.; Protopopova, M.N.; Winchester, W.P.; Tran, A. Ligand effects on dirhodium(II) carbene reactivities. Highly effective switch between competitive carbenoid transformations. J. Am. Chem. Soc. 1993, 115, 8669–8680. [Google Scholar] [CrossRef]
- Mbuvi, H.M.; Woo, L.K. Catalytic C-H Insertions Using Iron(III) Porphyrin Complexes. Organometallics 2008, 27, 637–645. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-M.; Cai, Y.; Zhu, S.-F.; Zhou, Q.-L. Iron-catalyzed arylation of α-aryl-α -diazoesters. Org. Biomol. Chem. 2016, 14, 5516–5519. [Google Scholar] [CrossRef]
- Wang, B.; Howard, I.G.; Pope, J.W.; Conte, E.D.; Deng, Y. Bis(imino)pyridine iron complexes for catalytic carbene transfer reactions. Chem. Sci. 2019, 10, 7958–7963. [Google Scholar] [CrossRef]
- Baumann, L.K.; Mbuvi, H.M.; Du, G.; Woo, L.K. Iron Porphyrin Catalyzed N-H Insertion Reactions with Ethyl Diazoacetate. Organometallics 2007, 26, 3995–4002. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Zhu, S.-F.; Wang, G.-P.; Zhou, Q.-L. Iron-Catalyzed C-H Functionalization of Indoles. Adv. Synth. Catal. 2011, 353, 2939–2944. [Google Scholar] [CrossRef]
- Hock, K.J.; Knorrscheidt, A.; Hommelsheim, R.; Ho, J.; Weissenborn, M.J.; Koenigs, R.M. Tryptamine Synthesis by Iron Porphyrin Catalyzed C-H Functionalization of Indoles with Diazoacetonitrile. Angew. Chem. Int. Ed. 2019, 58, 3630–3634. [Google Scholar] [CrossRef]
- Wang, B.; Qui, D.; Zhang, Y.; Wang, J. Recent advantages in C(sp3)-H bond functionalization via metal-carbene insertion. Beilstein J. Org. Chem. 2016, 12, 796–804. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Harmann, L.G.; Davies, H.M.L.; Beckwith, R.E.J. Late-stage C-H functionalization of complex alkaloids and drug molecules via intramolecular rhodium-carbenoid insertion. Nat. Commun. 2015, 6, 5943–5952. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.-Q.; Yang, J.-M.; Xu, H.; Zhu, S.-F. Iron-Catalyzed Carbenoid Insertion into C(sp3)-H Bonds. Synlett 2017, 28, 1327–1330. [Google Scholar]
- Griffin, J.R.; Wendell, C.I.; Garwin, J.A.; White, M.C. Catalytic C(sp3)-H Alkylation via an Iron Carbene Intermediate. J. Am. Chem. Soc. 2017, 139, 13624–13627. [Google Scholar] [CrossRef] [Green Version]
- Hernán-Gómez, A.; Rodriges, M.; Parella, T.; Costas, M. Electrophilic Iron Catalyst Paired with a Lithium Cation Enables Selective Functionalization of Non-Activated Aliphatic C-H Bonds via Metallocarbene Intermediates. Angew. Chem. Int. Ed. 2019, 58, 13904–13911. [Google Scholar] [CrossRef] [PubMed]
- Brandenberg, O.F.; Fasan, R.; Arnold, F.H. Exploiting and engineering hemoproteins for abiological carbene and nitrene transfer reactions. Curr. Opin. Biotechnol. 2017, 47, 102–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissenborn, M.J.; Koenigs, R.M. Iron-porphyrin catalyzed carbene transfer reactions—An evolution from biomimetic catalysis towards chemistry-inspired non-natural reactivities of enzymes. ChemCatChem 2019. [Google Scholar] [CrossRef]
- Vargas, D.V.; Tinoco, A.; Tyagi, V.; Fasan, R. Myoglobin-Catalyzed C-H Functionalization of Unprotected Indoles. Angew. Chem. Int. Ed. 2018, 57, 9911–9915. [Google Scholar] [CrossRef]
- Brandenberg, O.F.; Chen, K.; Arnold, F.H. Directed Evolution of a Cytochrome P450 Carbene Transferase for Selective Functionalization of Cyclic Compounds. J. Am. Chem. Soc. 2019, 141, 8989–8995. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.K.; Chen, K.; Huang, X.; Wohlschlager, L.; Renata, H.; Arnold, F.H. Enzymatic assembly of carbon-carbon bonds via iron-catalysed sp3 C-H functionalization. Nature 2019, 565, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Huang, X.; Zhang, R.K.; Arnold, F.H. Enantiodivergent α-Amino C-H Fluoroalkylation Catalysed by Engineered Cytochrome P450s. J. Am. Chem. Soc. 2019, 141, 9798–9802. [Google Scholar] [CrossRef] [Green Version]






















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Empel, C.; Jana, S.; Koenigs, R.M. C-H Functionalization via Iron-Catalyzed Carbene-Transfer Reactions. Molecules 2020, 25, 880. https://doi.org/10.3390/molecules25040880
Empel C, Jana S, Koenigs RM. C-H Functionalization via Iron-Catalyzed Carbene-Transfer Reactions. Molecules. 2020; 25(4):880. https://doi.org/10.3390/molecules25040880
Chicago/Turabian StyleEmpel, Claire, Sripati Jana, and Rene M. Koenigs. 2020. "C-H Functionalization via Iron-Catalyzed Carbene-Transfer Reactions" Molecules 25, no. 4: 880. https://doi.org/10.3390/molecules25040880
APA StyleEmpel, C., Jana, S., & Koenigs, R. M. (2020). C-H Functionalization via Iron-Catalyzed Carbene-Transfer Reactions. Molecules, 25(4), 880. https://doi.org/10.3390/molecules25040880