Candida antarctica Lipase A-Based Enantiorecognition of a Highly Strained 4-Dibenzocyclooctynol (DIBO) Used for PET Imaging

 , ,
, ,  ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
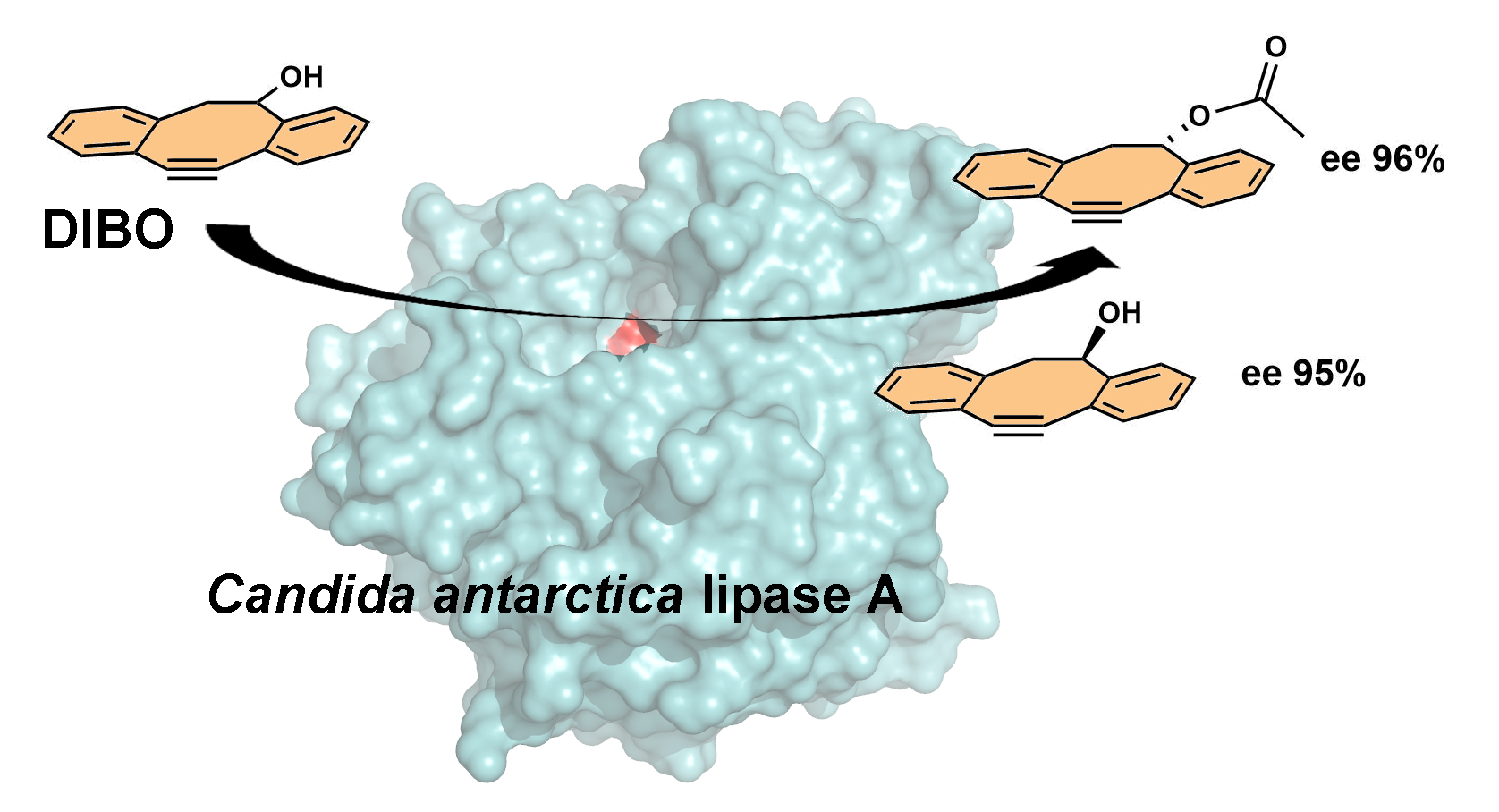
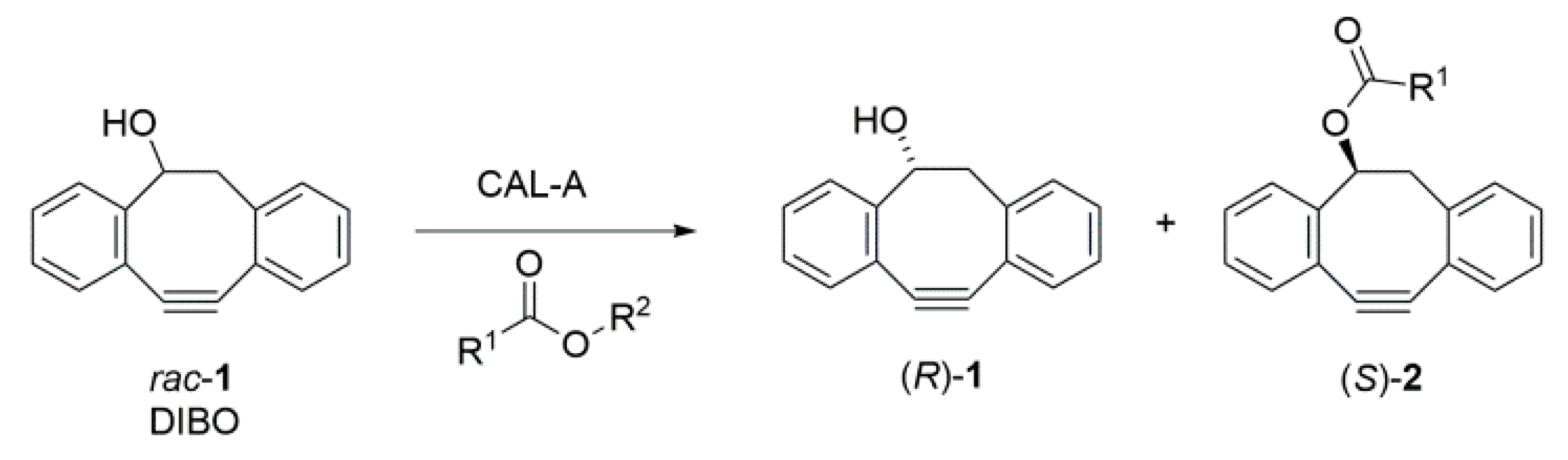
2.1. Enantiorecognition of DIBO by CAL-A
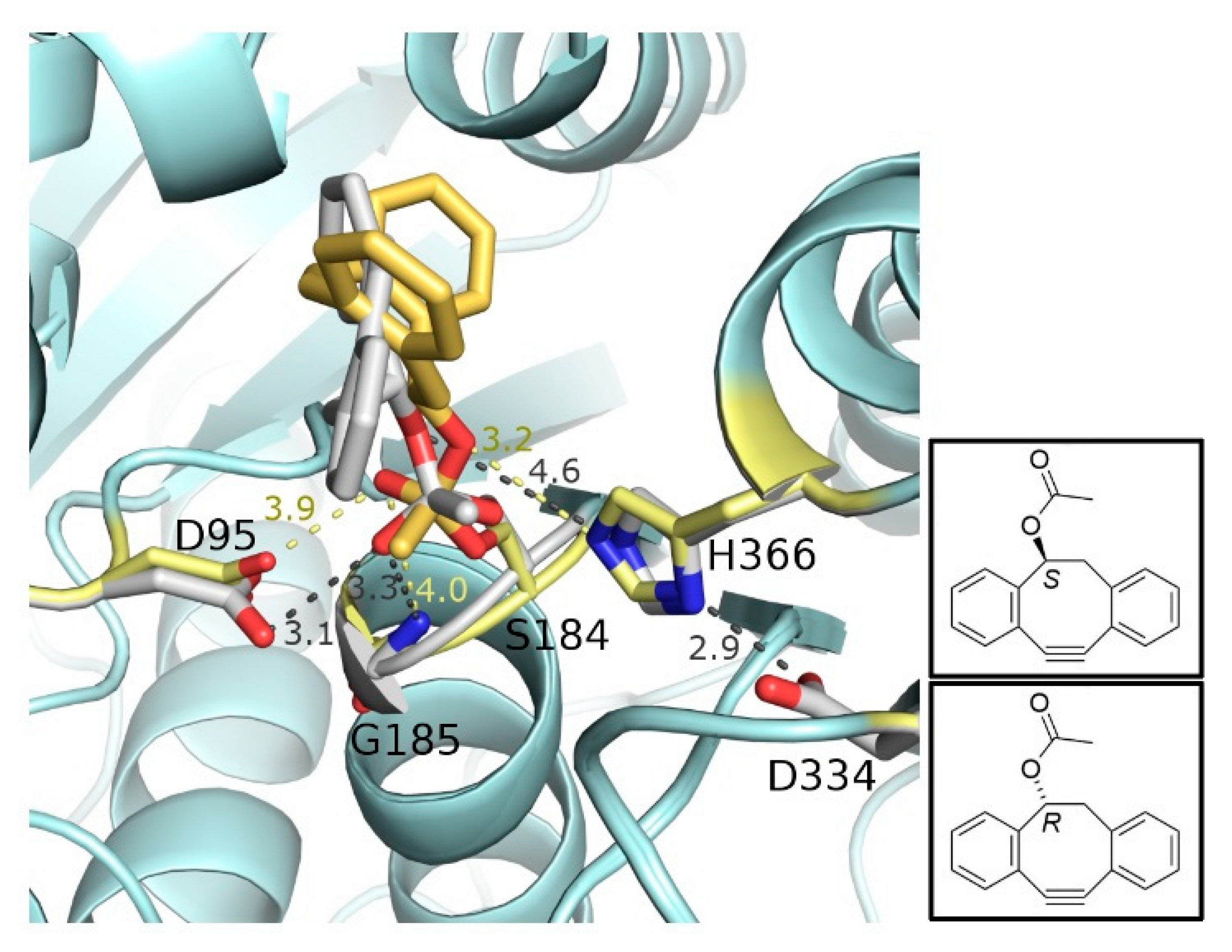
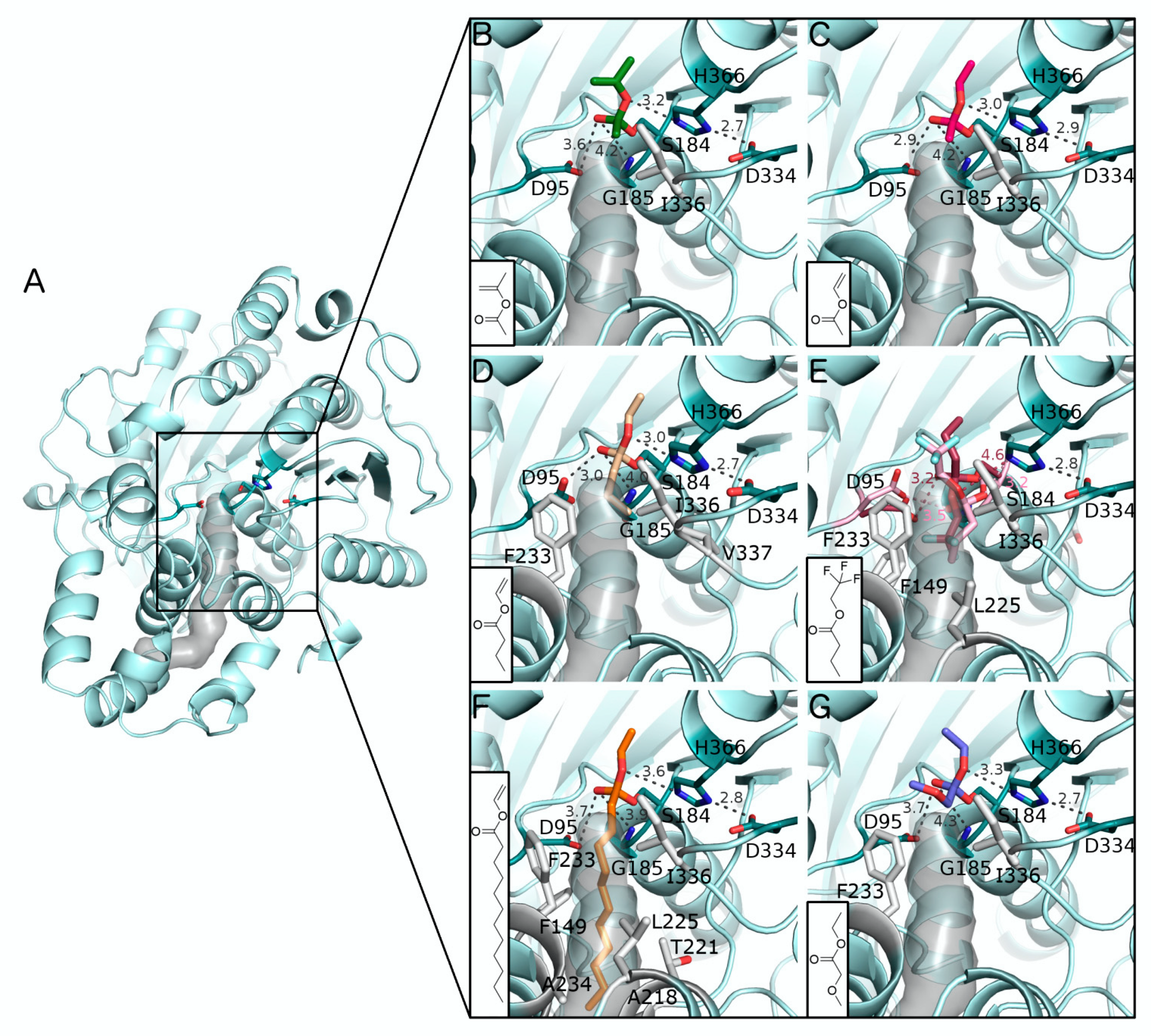
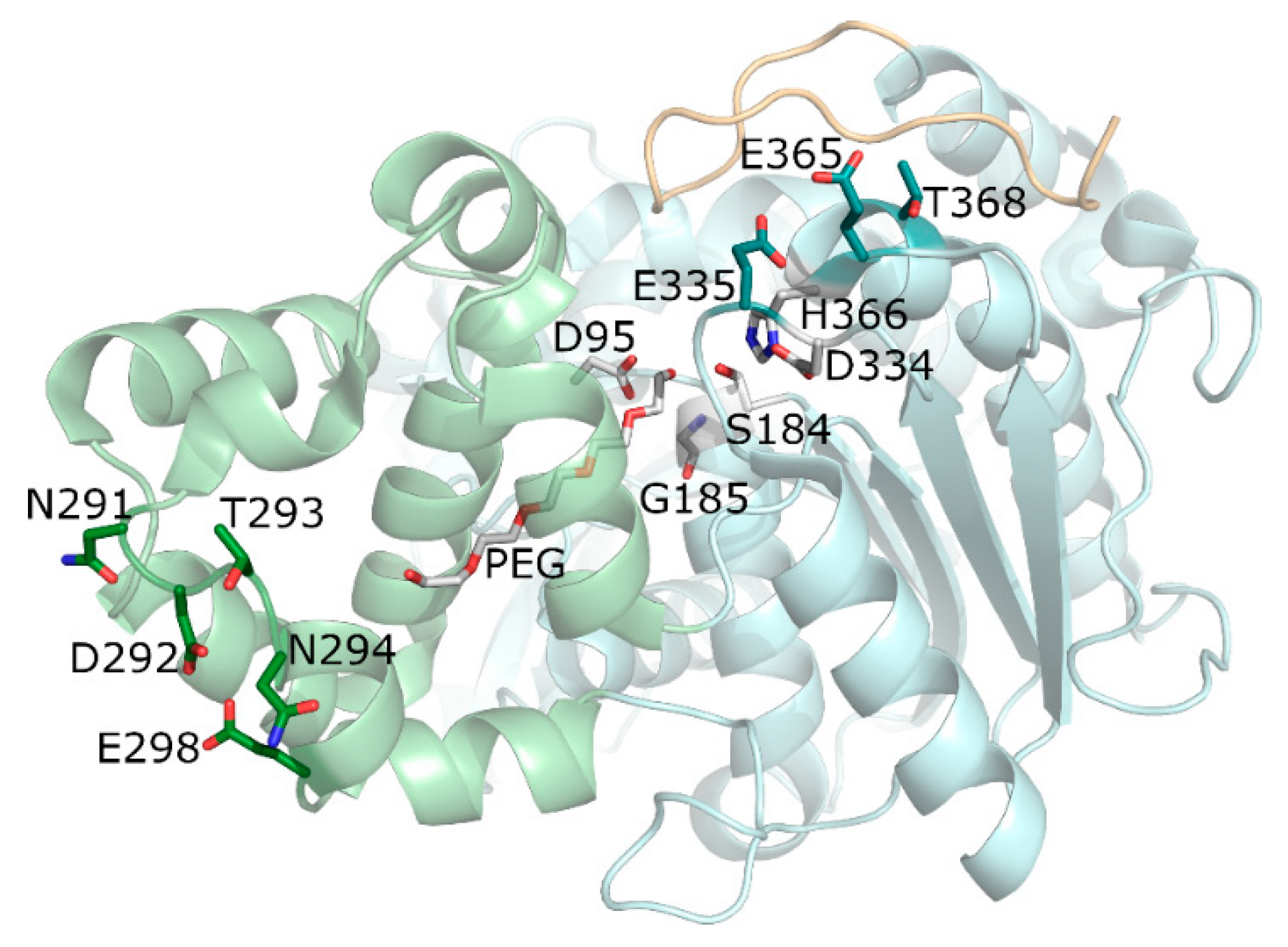
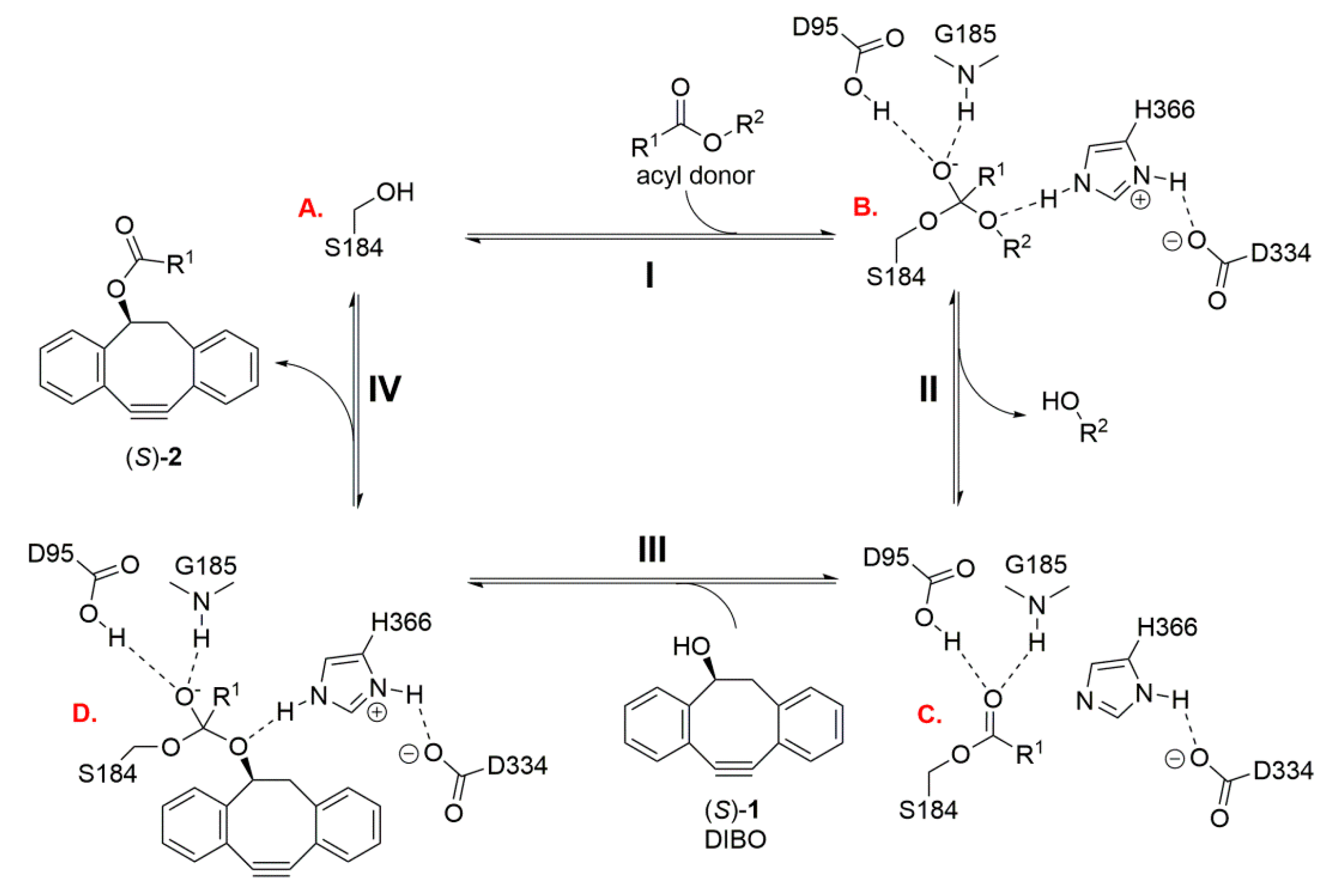
2.2. In Silico Docking of Acyl Donors
2.3. Effect of Drying Agents and Magnesium Ions
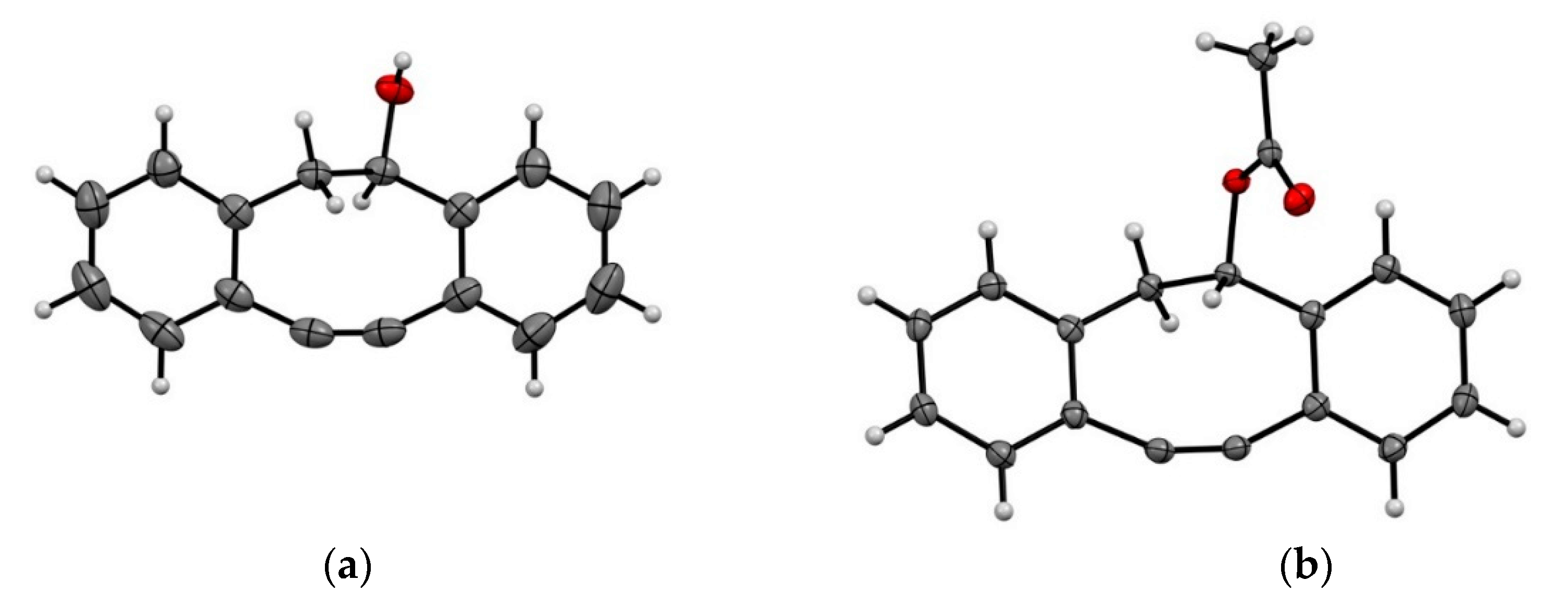
2.4. Separation of the Enantiomers and Determination of the Absolute Configurations
3. Materials and Methods
3.1. General Information
3.2. Instrumentation
3.3. Analytical Scale Reactions
3.4. Preparative Scale O-acylation of Rac-1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, X.-G.; Roivainen, A.; Bergman, J.; Heinonen, A.; Bengel, F.; Thum, T.; Knuuti, J. Enabling [18F]-bicyclo[6.1.0]nonyne for oligonucleotide conjugation for positron emission tomography applications: [18F]-anti-microRNA-21 as an example. J. Chem. Commun. 2015, 51, 9821–9824. [Google Scholar] [CrossRef]
- Li, X.-G.; Helariutta, K.; Roivainen, A.; Jalkanen, S.; Knuuti, J.; Airaksinen, A.J. Using 5-deoxy-5-[18F]fluororibose to glycosylate peptides for positron emission tomography. Nat. Protoc. 2014, 9, 138–145. [Google Scholar] [CrossRef]
- Knall, A.; Slugovc, C. Inverse electron demand Diels–Alder (iEDDA)-initiated conjugation: A (high) potential click chemistry scheme. Chem. Soc. Rev. 2013, 42, 5131–5142. [Google Scholar] [CrossRef] [Green Version]
- Houghton, J.L.; Zeglis, B.M.; Abdel-Atti, D.; Aggeler, R.; Sawada, R.; Agnew, B.J.; Scholz, W.W.; Lewis, J.S. Site-specifically labeled CA19.9-targeted immunoconjugates for the PET, NIRF, and multimodal PET/NIRF imaging of pancreatic cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15850–15855. [Google Scholar] [CrossRef] [Green Version]
- Buckingham, F.; Kirjavainen, A.K.; Forsback, S.; Krzyczmonik, A.; Keller, T.; Newington, I.M.; Glaser, M.; Luthra, S.K.; Solin, O.; Gouverneur, V. Organomediated Enantioselective 18F Fluorination for PET. Angew. Chem. Int. Ed. 2015, 54, 13366–13369. [Google Scholar] [CrossRef]
- Brem, J.; Liljeblad, A.; Paizs, C.; Tosa, M.I.; Irimie, F.; Kanerva, L.T. Lipases A and B from Candida antarctica in the enantioselective acylation of ethyl 3-heteroaryl-3-hydroxypropanoates: Aspects on the preparation and enantiopreference. Tetrahedron Asymmetry 2011, 22, 315–322. [Google Scholar] [CrossRef]
- Kim, S.; Choi, Y.K.; Hong, J.; Park, J.; Kim, M. Candida antarctica lipase A and Pseudomonas stutzeri lipase as a pair of stereocomplementary enzymes for the resolution of 1,2-diarylethanols and 1,2-diarylethanamines. Tetrahedron Lett. 2013, 54, 1185–1188. [Google Scholar] [CrossRef]
- Liljeblad, A.; Kanerva, L.T. Biocatalysis as a profound tool in the preparation of highly enantiopure β-amino acids. Tetrahedron 2006, 62, 5831–5854. [Google Scholar] [CrossRef]
- Gyarmati, Z.C.; Liljeblad, A.; Argay, G.; Kalman, A.; Bernath, G. Kanerva, L.T. Chemoenzymatic preparation of enantiopure homoadamantyl β-amino acid and β-lactam derivatives. Adv. Synth. Catal. 2004, 346, 566–572. [Google Scholar] [CrossRef]
- Ericsson, D.J.; Kasrayan, A.; Johansson, P.; Bergfors, T.; Sandström, A.G.; Bäckvall, J.; Mowbray, S.L. X-ray structure of Candida antarctica lipase A shows a novel lid structure and a likely mode of interfacial activation. J. Mol. Biol. 2008, 376, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikmark, Y.; Cassimjee, K.E.; Lihammar, R.; Bäckvall, J.-E. Removing the Active-Site Flap in lipase A from Candida antarctica Produces a Functional Enzyme without Interfacial Activation. ChemBioChem 2006, 17, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Özdemirhan, D.; Sezer, S.; Sönmez, Y. Enzyme-catalyzed resolution of aromatic ring fused cyclic tertiary alcohols. Tetrahedron Asymmetry 2008, 19, 2717–2720. [Google Scholar] [CrossRef]
- Erik, H.; Jürgen, P.; Bornscheuer, U.T. Activity of lipases and esterases towards tertiary alcohols: Insights into structure–function relationships. Angew. Chemie Int. Ed. 2002, 41, 3211–3213. [Google Scholar]
- Hietanen, A.; Lundell, K.; Kanerva, L.T.; Liljeblad, A. Advances in the kinetic and dynamic kinetic resolution of piperazine-2-carboxylic acid derivatives with Candida antarctica lipase A; structural requirements for enantioselective N-acylation. ARKIVOC 2012, 60–74. [Google Scholar] [CrossRef] [Green Version]
- Liljeblad, A.; Kiviniemi, A.; Kanerva, L.T. Aldehyde-based racemization in the dynamic kinetic resolution of N-heterocyclic α-amino esters using Candida antarctica lipase A. Tetrahedron 2004, 60, 671–677. [Google Scholar] [CrossRef]
- Gedey, S.; Lazar, L.; Fülöp, F.; Liljeblad, A.; Kanerva, L.T. Preparation of highly enantiopure β-amino esters by Candida antarctica lipase A. Tetrahedron Asymmetry 2001, 15, 105–110. [Google Scholar] [CrossRef]
- Hanefeld, U. Reagents for (ir)reversible enzymatic acylations. Org. Biomol. Chem. 2003, 1, 2405–2415. [Google Scholar] [CrossRef]
- Brandt, A.-M.; Li, X.-G.; Nymalm-Rejstrom, Y.; Airenne, T.; Kanerva, L.T.; Salminen, T.A. The crystal structure of lipase A from Candida antarctica. RCSB Protein Data Bank 2009. [Google Scholar] [CrossRef]
- Mäenpää, H.; Kanerva, L.T.; Liljeblad, A. Acylation of β-Amino Esters and Hydrolysis of β-Amido Esters: Candida antarctica Lipase A as a Chemoselective Deprotection Catalyst. ChemCatChem 2016, 8, 1226–1232. [Google Scholar] [CrossRef]
- Sundell, R.; Siirola, E.; Kanerva, L.T. Regio- and Stereoselective Lipase-Catalysed Acylation of Methyl α-d-Glycopyranosides with Fluorinated β-Lactams. Eur. J. Org. Chem. 2014, 2014, 6753–6760. [Google Scholar] [CrossRef]
- Kvittingen, L.; Sjursnes, B.; Anthonsen, T.; Halling, P. Use of salt hydrates to buffer optimal water level during lipase catalysed in synthesis in organic media: A practical procedure for organic chemists. Tetrahedron 1992, 48, 2793–2802. [Google Scholar] [CrossRef]
- Lu, C.-H.; Lin, Y.-F.; Lin, J.-J.; Yu, C.-S. Prediction of metal ion–binding sites in proteins using the fragment transformation method. PLoS ONE 2012, 7, e39252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flack, H.D.; Bernardinelli, G. The use of X-ray crystallography to determine absolute configuration. Chirality 2008, 20, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Flack, H.D. Chiral and achiral crystal structures. Helv. Chim. Acta 2003, 86, 905–921. [Google Scholar] [CrossRef]
- Chen, C.S.; Fujimoto, Y.; Girdaukas, G.; Sih, C. Quantitative analyses of biochemical kinetic resolutions of enantiomers. J. Am. Chem. Soc. 1982, 104, 7294–7299. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Acyl Donor | R1 | R2 | ees (%) | eep (%) | c (%) | Ea) |
|---|---|---|---|---|---|---|---|
| 1 | Isopropenyl acetate | Me | C(CH3)=CH2 | 98 | 96 | 51 | 240 |
| 2 | Vinyl acetate | Me | CH=CH2 | 91 | 95 | 49 | 130 |
| 3 | Ethyl acetate | Me | Et | 7 | >99 | 7 | -b) |
| 4 | Vinyl butanoate | Pr | CH=CH2 | 18 | 98 | 15 | 90 |
| 5 | Trifluoroethyl butanoate | Pr | CH2CF3 | 19 | >99 | 16 | >200 |
| 6 | Vinyl laurate | CH3(CH2)10 | CH=CH2 | 8 | 96 | 7 | -b) |
| 7 | Ethyl methoxyacetate | MeOCH2 | Et | 0 | - | 0 | - |
| Entry | Acyl Donor | Additives | t (h) | ees (%) | eep (%) | c (%) | E |
|---|---|---|---|---|---|---|---|
| 1 | Isopropenyl acetate | none | 24 | 90 | 97 | 48 | 240 |
| 2 | Isopropenyl acetate | none | 96 | 98 | 96 | 51 | 240 |
| 3 | Isopropenyl acetate | 3 Å molecular sieves a) | 24 | 97 | 94 | 51 | 185 |
| 4 | Isopropenyl acetate | MgSO4 (anh.) | 24 | 95 | 96 | 50 | 185 |
| 5 | Isopropenyl acetate | MgCl2 (anh.) | 24 | 93 | 97 | 49 | 210 |
| 6 | Isopropenyl acetate | MgCl2·6H2O | 24 | 86 | 99 | 47 | 355 |
| 7 | Vinyl acetate | none | 96 | 91 | 95 | 49 | 130 |
| 8 | Vinyl acetate | MgCl2 (anh.) | 24 | 96 | 93 | 51 | 95 |
| 9 | Vinyl acetate | MgCl2·6H2O | 24 | 96 | 95 | 50 | 135 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirén, S.; Dahlström, K.M.; Puttreddy, R.; Rissanen, K.; Salminen, T.A.; Scheinin, M.; Li, X.-G.; Liljeblad, A. Candida antarctica Lipase A-Based Enantiorecognition of a Highly Strained 4-Dibenzocyclooctynol (DIBO) Used for PET Imaging. Molecules 2020, 25, 879. https://doi.org/10.3390/molecules25040879
Sirén S, Dahlström KM, Puttreddy R, Rissanen K, Salminen TA, Scheinin M, Li X-G, Liljeblad A. Candida antarctica Lipase A-Based Enantiorecognition of a Highly Strained 4-Dibenzocyclooctynol (DIBO) Used for PET Imaging. Molecules. 2020; 25(4):879. https://doi.org/10.3390/molecules25040879
Chicago/Turabian StyleSirén, Saija, Käthe M. Dahlström, Rakesh Puttreddy, Kari Rissanen, Tiina A. Salminen, Mika Scheinin, Xiang-Guo Li, and Arto Liljeblad. 2020. "Candida antarctica Lipase A-Based Enantiorecognition of a Highly Strained 4-Dibenzocyclooctynol (DIBO) Used for PET Imaging" Molecules 25, no. 4: 879. https://doi.org/10.3390/molecules25040879
APA StyleSirén, S., Dahlström, K. M., Puttreddy, R., Rissanen, K., Salminen, T. A., Scheinin, M., Li, X. -G., & Liljeblad, A. (2020). Candida antarctica Lipase A-Based Enantiorecognition of a Highly Strained 4-Dibenzocyclooctynol (DIBO) Used for PET Imaging. Molecules, 25(4), 879. https://doi.org/10.3390/molecules25040879