
Enantioselective Iron/Bisquinolyldiamine Ligand-Catalyzed Oxidative Coupling Reaction of 2-Naphthols


Abstract
:
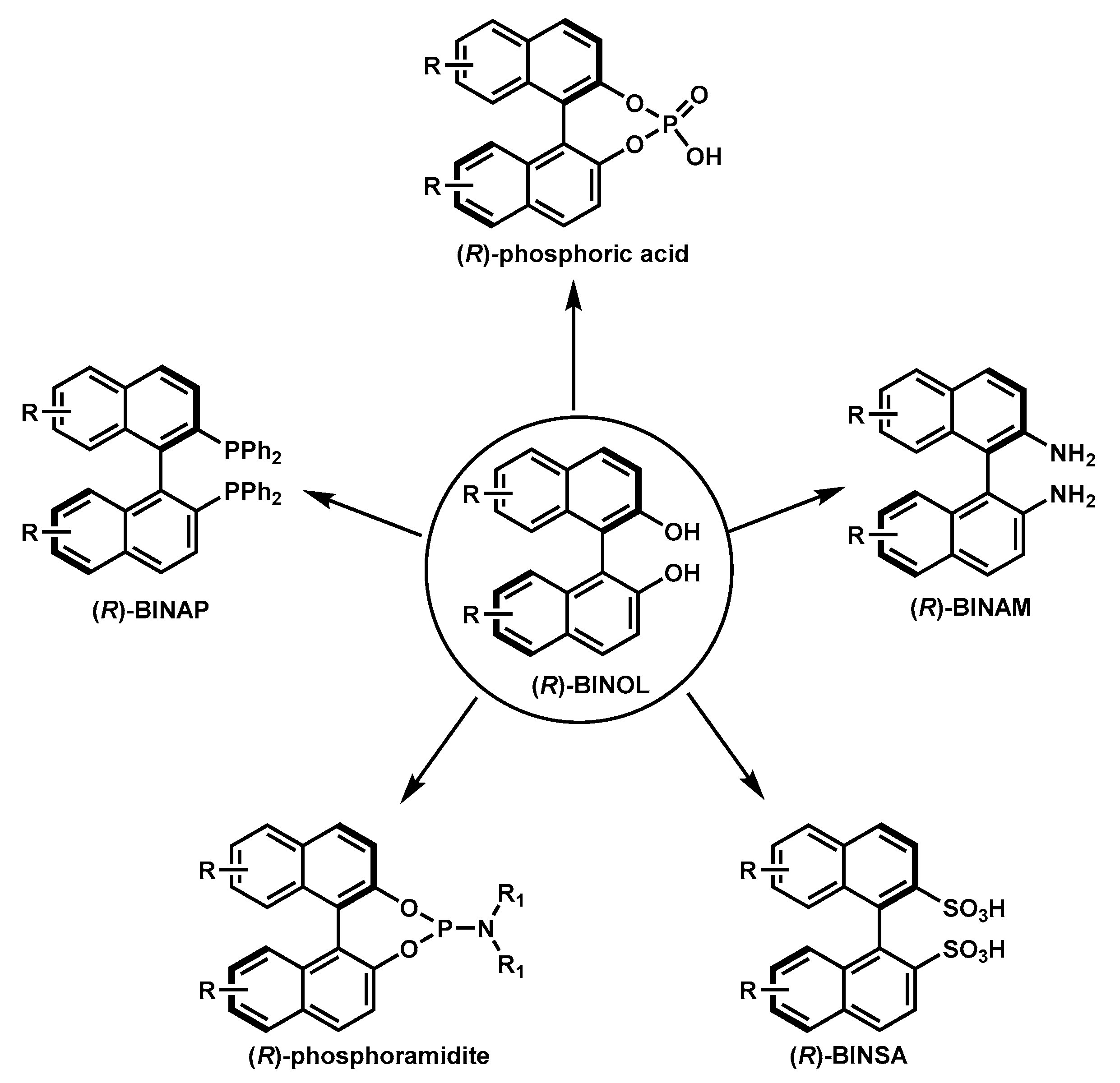
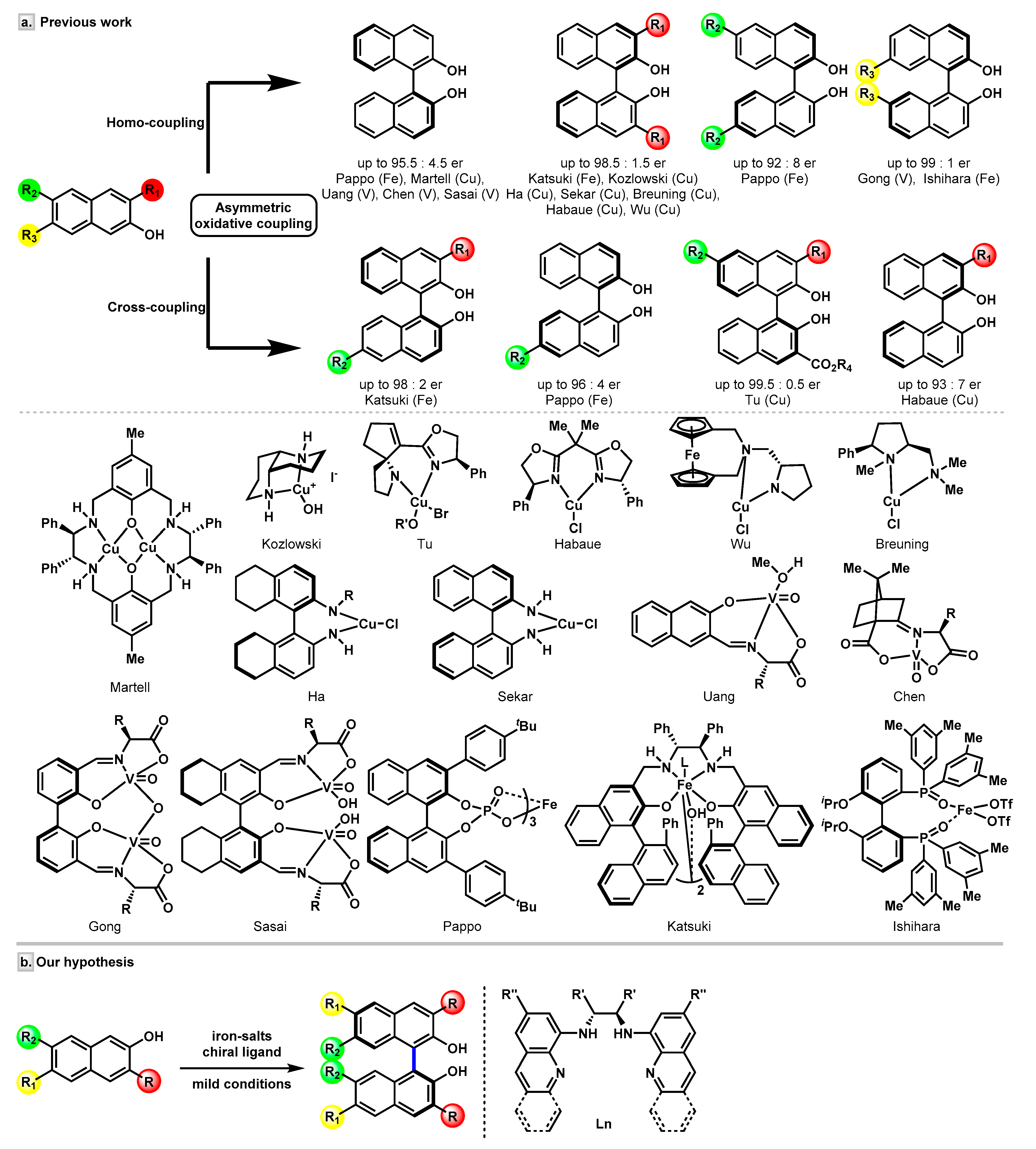
1. Introduction
2. Results and Discussion
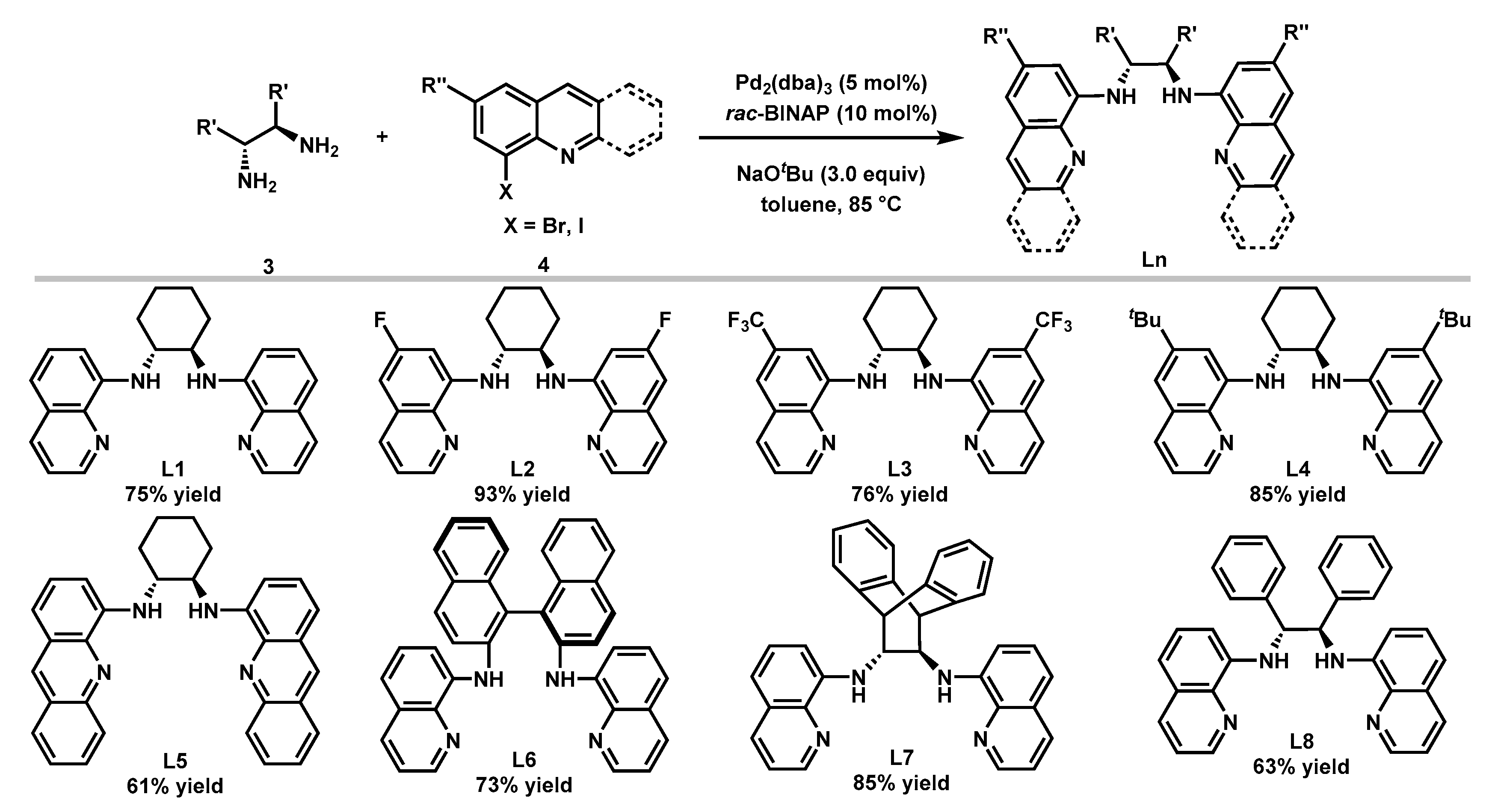
2.1. Synthesis of Bisquinolyldiamine Ligands
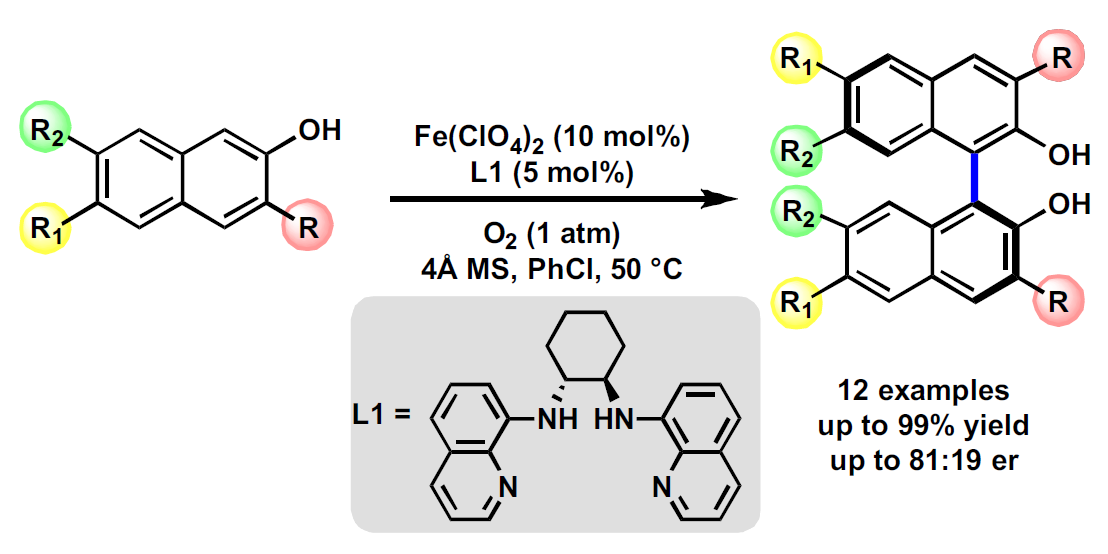
2.2. Reaction Investigation
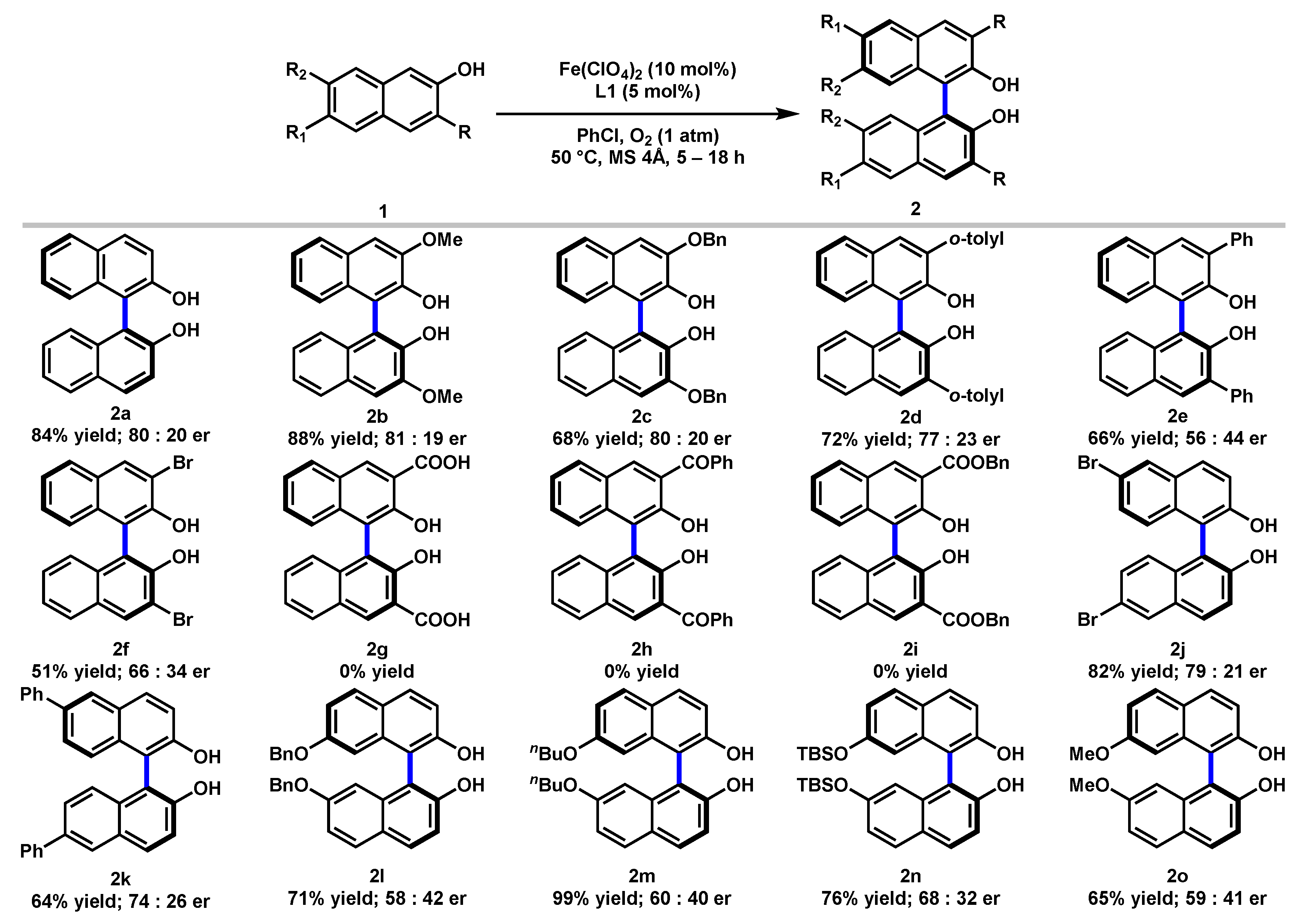
2.3. Substrates Scope
3. Materials and Methods
3.1. General Information
3.2. Preparation of Ligands
3.3. Preparation of Substituted Quinolines
3.4. Iron-Catalyzed Asymmetric Oxidative Coupling Reaction of 2-Naphthols
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, Y.-B.; Tan, B. Construction of Axially Chiral Compounds via Asymmetric Organocatalysis. Acc. Chem. Res. 2018, 51, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Wencel-Delord, J.; Panossian, A.; Leroux, F.R.; Colobert, F. Recent Advances and New Concepts for the Synthesis of Axially Stereoenriched Biaryls. Chem. Soc. Rev. 2015, 44, 3418–3430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moliterno, M.; Cari, R.; Puglisi, A.; Antenucci, A.; Sperandio, C.; Moretti, E.; Sabato, A.D.; Salvio, R.; Bella, M. Quinine-Catalyzed Asymmetric Synthesis of 2,2′-Binaphthol-type Biaryls under Mild Reaction Conditions. Angew. Chem. Int. Ed. 2016, 128, 6635–6639. [Google Scholar] [CrossRef]
- Wang, J.-Z.; Zhou, J.; Xu, C.; Sun, H.; Kürti, L.; Xu, Q.-L. Symmetry in Cascade Chirality-Transfer Processes: A Catalytic Atroposelective Direct Arylation Approach to BINOL Derivatives. J. Am. Chem. Soc. 2016, 138, 5202–5205. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-H.; Cheng, D.-J.; Zhang, J.; Wang, Y.; Liu, X.-Y.; Tan, B. Atroposelective Synthesis of Axially Chiral Biaryldiols via Organocatalytic Arylation of 2-Naphthols. J. Am. Chem. Soc. 2015, 137, 15062–15065. [Google Scholar] [CrossRef]
- Wang, H. Recent Advances in Asymmetric Oxidative Coupling of 2-Naphthol and Its Derivatives. Chirality 2010, 22, 827–837. [Google Scholar] [CrossRef]
- Brunel, J.M. BINOL: A Versatile Chiral Reagent. Chem. Rev. 2005, 105, 857–898. [Google Scholar] [CrossRef]
- Chen, Y.; Yekta, S.; Yudin, A.K. Modified BINOL Ligands in Asymmetric Catalysis. Chem. Rev. 2003, 103, 3155–3212. [Google Scholar] [CrossRef]
- Pu, L. 1,1′-Binaphthyl Dimers, Oligomers, and Polymers: Molecular Recognition, Asymmetric Catalysis, and New Materials. Chem. Rev. 1998, 98, 2405–2494. [Google Scholar] [CrossRef]
- Tomino, I.; Tanimoto, Y.; Noyori, R. Virtually Complete Enantioface Differentiation in Carbonyl Group Reduction by a Complex Aluminum Hydride Reagent. J. Am. Chem. Soc. 1979, 101, 3129–3131. [Google Scholar]
- Berthod, M.; Mignani, G.; Woodward, G.; Lemaire, M. Modified BINAP: The How and the Why. Chem. Rev. 2005, 105, 1801–1836. [Google Scholar] [CrossRef] [PubMed]
- Kočovský, P.; Vyskočil, Š.; Smrčina, M. Non-Symmetrically Substituted 1,1′-Binaphthyls in Enantioselective Catalysis. Chem. Rev. 2003, 103, 3213–3245. [Google Scholar] [CrossRef] [PubMed]
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev. 2014, 114, 9047–9153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-F.; Xie, F.; Yang, B.; Yu, H.; Zhang, W.-B. Chiral Phosphoramidite Ligand and Its Application in Asymmetric Catalysis. Chin. J. Org. Chem. 2011, 31, 429–442. [Google Scholar]
- Hatano, M.; Ishihara, K. Chiral 1,1′-Binaphthyl-2,2′-Disulfonic Acid (BINSA) and Its Derivatives for Asymmetric Catalysis. Asian J. Org. Chem. 2014, 3, 352–365. [Google Scholar] [CrossRef]
- Komanduri, V.; Krische, M.J. Enantioselective Reductive Coupling of 1,3-Enynes to Heterocyclic Aromatic Aldehydes and Ketones via Rhodium-Catalyzed Asymmetric Hydrogenation: Mechanistic Insight into the Role of Brønsted Acid Additives. J. Am. Chem. Soc. 2006, 128, 16448–16449. [Google Scholar] [CrossRef]
- Costa, A.L.; Piazza, M.G.; Tagliavini, E.; Trombini, C.; Ronchi, A.U. Catalytic Asymmetric Synthesis of Homoallylic Alcohols. J. Am. Chem. Soc. 1993, 115, 7001–7002. [Google Scholar] [CrossRef]
- Sakane, S.; Fujiwara, J.; Maruoka, K.; Yamamoto, H. Chiral Leaving Group. Biogenetic-Type Asymmetric Synthesis of Limonene and Bisabolenes. J. Am. Chem. Soc. 1983, 105, 6154–6155. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Inubushi, A.; Suda, S.; Hara, R.; Kobayashi, S. [1,1′-Bi-2-naphthalenediolato(2-)-O,O′]oxotitanium. An Efficient Chiral Catalyst for the Asymmetric Aldol Reaction of Silyl Enol Ethers with Aldehydes. Chem. Lett. 1990, 19, 1015–1018. [Google Scholar] [CrossRef]
- Qi, L.-W.; Mao, J.-H.; Zhang, J.; Tan, B. Organocatalytic Asymmetric Arylation of Indoles Enabled by Azo Groups. Nat. Chem. 2018, 10, 58–64. [Google Scholar] [CrossRef]
- Takizawa, S. Development of Dinuclear Vanadium Catalysts for Enantioselective Coupling of 2-Naphthols via a Dual Activation Mechanism. Chem. Pharm. Bull. 2009, 57, 1179–1188. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.-Y.; Hwang, D.R.; Wang, S.-K.; Uang, B.J. Chiral Oxovanadium Complex Catalyzed Enantioselective Oxidative Coupling of 2-Naphthols. Chem. Commun. 2001, 37, 980–981. [Google Scholar] [CrossRef] [Green Version]
- Barhate, N.B.; Chen, C.-T. Catalytic Asymmetric Oxidative Couplings of 2-Naphthols by Tridentate N-Ketopinidene-Based Vanadyl Dicarboxylates. Org. Lett. 2002, 4, 2529–2532. [Google Scholar] [CrossRef]
- Guo, Q.-X.; Wu, Z.-J.; Luo, Z.-B.; Liu, Q.-Z.; Ye, J.-L.; Luo, S.-W.; Cun, L.-F.; Gong, L.-Z. Highly Enantioselective Oxidative Couplings of 2-Naphthols Catalyzed by Chiral Bimetallic Oxovanadium Complexes with either Oxygen or Air as Oxidant. J. Am. Chem. Soc. 2007, 129, 13927–13938. [Google Scholar] [CrossRef]
- Luo, Z.; Liu, Q.; Gong, L.-Z.; Cui, X.; Mi, A.; Jiang, Y. Novel Achiral Biphenol-Derived Diastereomeric Oxovanadium(IV) Complexes for Highly Enantioselective Oxidative Coupling of 2-Naphthols. Angew. Chem. Int. Ed. 2002, 41, 4532–4535. [Google Scholar] [CrossRef]
- Luo, Z.-B.; Liu, Q.-Z.; Gong, L.-Z. The Rational Design of Novel Chiral Oxovanadium(IV) Complexes for Highly Enantioselective Oxidative Coupling of 2-Naphthols. Chem. Commun. 2002, 38, 914–915. [Google Scholar] [CrossRef] [PubMed]
- Sako, M.; Takizawa, S.; Yoshida, Y.; Sasai, H. Enantioselective and Aerobic Oxidative Coupling of 2-Naphthol Derivatives Using Chiral Dinuclear Vanadium(V) Complex in Water. Tetrahedron Asymmetry 2015, 26, 613–616. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, S.; Katayama, T.; Sasai, H. Dinuclear Chiral Vanadium Catalysts for Oxidative Coupling of 2-Naphtholsvia a Dual Activation Mechanism. Chem. Commun. 2008, 44, 4113–4122. [Google Scholar] [CrossRef]
- Takizawa, S.; Katayama, T.; Somei, H.; Asano, Y.; Yoshida, T.; Kameyama, C.; Rajesh, D.; Onitsuka, K.; Suzuki, T.; Mikami, M.; et al. Dual Activation in Oxidative Coupling of 2-Naphthols Catalyzed by Chiral Dinuclear Vanadium Complexes. Tetrahedron 2008, 64, 3361–3371. [Google Scholar] [CrossRef]
- Nakajima, M.; Miyoshi, I.; Kanayama, K.; Hashimoto, S.; Noji, M.; Koga, K. Enantioselective Synthesis of Binaphthol Derivatives by Oxidative Coupling of Naphthol Derivatives Catalyzed by Chiral Diamine•Copper Complexes. J. Org. Chem. 1999, 64, 2264–2271. [Google Scholar] [CrossRef]
- Gao, J.; Reibenspies, J.H.; Martell, A.E. Structurally Defined Catalysts for Enantioselective Oxidative Coupling Reactions. Angew. Chem. Int. Ed. 2003, 42, 6008–6012. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, M.C.; Morgan, B.J.; Linton, E.C. Total Synthesis of Chiral Biaryl Natural Products by Asymmetric Biaryl Coupling. Chem. Soc. Rev. 2009, 38, 3193–3207. [Google Scholar] [CrossRef] [PubMed]
- Hewgley, J.B.; Stahl, S.S.; Kozlowski, M.C. Mechanistic Study of Asymmetric Oxidative Biaryl Coupling: Evidence for Self-Processing of the Copper Catalyst to Achieve Control of Oxidase vs. Oxygenase Activity. J. Am. Chem. Soc. 2008, 130, 12232–12233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podlesny, E.E.; Kozlowski, M.C. Enantioselective Total Synthesis of (S)-Bisoranjidiol, an Axially Chiral Bisanthraquinone. Org. Lett. 2012, 14, 1408–1411. [Google Scholar] [CrossRef]
- O’Brien, E.M.; Morgan, B.J.; Mulrooney, C.A.; Carroll, P.J.; Kozlowski, M.C. Perylenequinone Natural Products: Total Synthesis of Hypocrellin A. J. Org. Chem. 2010, 75, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Hewgley, J.B.; Mulrooney, C.A.; Yang, J.; Kozlowski, M.C. Enantioselective Oxidative Biaryl Coupling Reactions Catalyzed by 1,5-Diazadecalin Metal Complexes: Efficient Formation of Chiral Functionalized BINOL Derivatives. J. Org. Chem. 2003, 68, 5500–5511. [Google Scholar] [CrossRef]
- Mulrooney, C.A.; Li, X.; DiVirgilio, E.S.; Kozlowski, M.C. General Approach for the Synthesis of Chiral Perylenequinones via Catalytic Enantioselective Oxidative Biaryl Coupling. J. Am. Chem. Soc. 2003, 125, 6856–6857. [Google Scholar] [CrossRef]
- Li, X.; Yang, J.; Kozlowski, M.C. Enantioselective Oxidative Biaryl Coupling Reactions Catalyzed by1,5-Diazadecalin Metal Complexes. Org. Lett. 2001, 3, 1137–1140. [Google Scholar] [CrossRef]
- Temma, T.; Hatano, B.; Habaue, S. Cu(I)-Catalyzed Asymmetric Oxidative Cross-Coupling of 2-Naphthol Derivatives. Tetrahedron 2006, 62, 8559–8563. [Google Scholar] [CrossRef]
- Temma, T.; Habaue, S. Highly Selective Oxidative Cross-Coupling of 2-Naphthol Derivatives with Chiral Copper(I)–Bisoxazoline Catalysts. Tetrahedron Lett. 2005, 46, 5655–5657. [Google Scholar] [CrossRef]
- Prause, F.; Arensmeyer, B.; Fröhlich, B.; Breuning, M. In-Depth Structure–Selectivity Investigations on Asymmetric, Copper-Catalyzed Oxidative Biaryl Coupling in the Presence of 5-cis-Substituted Prolinamines. Catal. Sci. Technol. 2015, 5, 2215–2226. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-H.; Lee, D.-W.; Lee, Y.-S.; Ko, D.-H.; Ha, D.-C. Enantioselective Oxidative Coupling of Methyl 3-Hydroxy-2-naphthoate Using Mono-N-alkylated Octahydrobinaphthyl-2,2′-Diamine Ligand. Tetrahedron 2004, 60, 9037–9042. [Google Scholar] [CrossRef]
- Alamsetti, S.K.; Poonguzhali, E.; Ganapathy, D.; Sekar, G. Enantioselective Oxidative Coupling of 2-Naphthol Derivatives by Copper-(R)-1,1′-Binaphthyl-2,2′-Diamine-TEMPO Catalyst. Adv. Synth. Catal. 2013, 355, 2803–2808. [Google Scholar] [CrossRef]
- Zhang, Q.; Cui, X.; Chen, L.; Liu, H.; Wu, Y. Syntheses of Chiral Ferrocenophanes and Their Application to Asymmetric Catalysis. Eur. J. Org. Chem. 2014, 2014, 7823–7829. [Google Scholar] [CrossRef]
- Tian, J.-M.; Wang, A.-F.; Yang, J.-S.; Zhao, X.-J.; Tu, Y.-Q.; Zhang, S.-Y.; Chen, Z.-M. Copper-Complex-Catalyzed Asymmetric Aerobic Oxidative Cross-Coupling of 2-Naphthols: Enantioselective Synthesis of 3,3′-Substituted C1-Symmetric BINOLs. Angew. Chem. Int. Ed. 2019, 58, 11023–11027. [Google Scholar] [CrossRef] [PubMed]
- Egami, H.; Katsuki, T. Iron-Catalyzed Asymmetric Aerobic Oxidation: Oxidative Coupling of 2-Naphthols. J. Am. Chem. Soc. 2009, 131, 6082–6083. [Google Scholar] [CrossRef] [PubMed]
- Egami, H.; Matsumoto, K.; Oguma, T.; Kunisu, T.; Katsuki, T. Enantioenriched Synthesis of C1-Symmetric BINOLs: Iron-Catalyzed Cross-Coupling of 2-Naphthols and Some Mechanistic Insight. J. Am. Chem. Soc. 2010, 132, 13633–13635. [Google Scholar] [CrossRef]
- Horibe, T.; Nakagawa, K.; Hazeyama, T.; Takeda, K.; Ishihara, K. An Enantioselective Oxidative Coupling Reaction of 2-Naphthol Derivatives Catalyzed by Chiral Diphosphine Oxide–iron(II) Complexes. Chem. Commun. 2019, 55, 13677–13680. [Google Scholar] [CrossRef]
- Narute, S.; Parnes, R.; Toste, F.D.; Pappo, D. Enantioselective Oxidative Homocoupling and Cross-Coupling of 2-Naphthols Catalyzed by Chiral Iron Phosphate Complexes. J. Am. Chem. Soc. 2016, 138, 16553–16560. [Google Scholar] [CrossRef] [Green Version]
- Shalit, H.; Dyadyuk, A.; Pappo, D. Selective Oxidative Phenol Coupling by Iron Catalysis. J. Org. Chem. 2019, 84, 1677–1686. [Google Scholar] [CrossRef]
- Liu, W.; Zhong, D.-Y.; Yu, C.-L.; Zhang, Y.; Wu, D.; Feng, Y.-L.; Cong, H.-J.; Lu, X.-Q.; Liu, W.-B. Iron-Catalyzed Intramolecular Amination of Aliphatic C–H Bonds of Sulfamate Esters with High Reactivity and Chemoselectivity. Org. Lett. 2019, 21, 2673–2678. [Google Scholar] [CrossRef] [PubMed]
- Zhong, D.-Y.; Wu, D.; Zhang, Y.; Lu, Z.-W.; Usman, M.; Liu, W.; Lu, X.-Q.; Liu, W.-B. Synthesis of Sultams and Cyclic N-Sulfonyl Ketimines via Iron-Catalyzed Intramolecular Aliphatic C-H Amidation. Org. Lett. 2019, 21, 5808–5812. [Google Scholar] [CrossRef] [PubMed]
- Zang, C.; Liu, Y.-G.; Xu, Z.-J.; Tse, C.-T.; Guan, X.-G.; Wei, J.-H.; Huang, J.-S.; Che, C.-M. Highly Enantioselective Iron-Catalyzed cis-Dihydroxylation of Alkenes with Hydrogen Peroxide Oxidant via an Fe(III)-OOH Reactive Intermediate. Angew. Chem. Int. Ed. 2016, 55, 10253–10257. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.-H.; Cao, B.; Tse, C.-W.; Chang, X.-Y.; Zhou, C.-Y.; Che, C.-M. Chiral cis-Iron(II) Complexes with Metal- and Ligand-Centered Chirality for Highly Regio- and Enantioselective Alkylation of N-heteroaromatics. Chem. Sci. 2020, 11, 684–693. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef]
- Baumgartner, L.M.; Dennis, J.M.; White, N.A.; Buchwald, S.L.; Jensen, K.F. Use of a Droplet Platform to Optimize Pd-Catalyzed C–N Coupling Reactions Promoted by Organic Bases. Org. Process Res. Dev. 2019, 23, 1594–1601. [Google Scholar] [CrossRef]
- Pangborn, A.B.; Giardello, M.A.; Grubbs, R.H.; Rosen, R.K.; Timmers, F.J. Safe and Convenient Procedure for Solvent Purification. Organometallics 1996, 15, 1518–1520. [Google Scholar] [CrossRef]
- Michon, C.; Ellern, A.; Angelici, R.J. Chiral Tetradentate Amine and Tridentate Aminocarbene Ligands: Synthesis, Reactivity and X-Ray Structural Characterizations. Inorganica Chimica Acta 2006, 39, 4549–4556. [Google Scholar] [CrossRef]
- Ramann, G.A.; Cowen, B.J. Quinoline Synthesis by Improved Skraup–Doebner–Von Miller Reactions Utilizing Acrolein Diethyl Acetal. Tetrahedron Lett. 2015, 56, 6436–6439. [Google Scholar] [CrossRef]
- Chou, C.-C.; Hu, F.-C.; Yeh, H.-H.; Wu, H.-P.; Chi, Y.; Clifford, J.N.; Palomares, E.; Liu, S.-H.; Chou, P.-T.; Lee, G.-H. Highly Efficient Dye-Sensitized Solar Cells Based on Panchromatic Ruthenium Sensitizers with Quinolinyl Bipyridine Anchors. Angew. Chem. Int. Ed. 2014, 53, 178–183. [Google Scholar] [CrossRef]
- Patra, T.; Agasti, S.; Akanksha; Maiti, D. Nickel-Catalyzed Decyanation of Inert Carbon–Cyano Bonds. Chem. Commun. 2013, 49, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Niimi, K.; Mori, H.; Miyazaki, E.; Osaka, I.; Kakizoe, H.; Takimiya, K.; Adachi, C. [2,2ʹ] Bi[Naphtho [2,3-B] Furanyl]: A Versatile Organic Semiconductor with a Furan–Furan Junction. Chem. Commun. 2012, 48, 5892–5894. [Google Scholar] [CrossRef] [PubMed]
- Bolchi, C.; Catalano, P.; Fumagalli, L.; Gobbi, M.; Pallavicini, M.; Pedretti, A.; Villa, L.; Vistoli, G.; Valoti, E. Structure–Affinity Studies for a Novel Series of Homochiral Naphthol and Tetrahydronaphthol Analogues of A1 Antagonist WB-4101. Bioorganic Med. Chem. 2004, 12, 4937–4951. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Trzoss, M.; Brimble, M.A. A Facile Synthesis of Aryl Spirodioxines Based on a 3H,3ʹH-2,2ʹ-Spirobi (Benzo [b][1,4] Dioxine) Skeleton. Synthesis 2007, 9, 1392–1402. [Google Scholar]
- Forkosh, H.; Vershinin, V.; Reiss, H.; Pappo, D. Stereoselective Synthesis of Optically Pure 2-Amino-2′-Hydroxy-1,1′-Binaphthyls. Org. Lett. 2018, 20, 2459–2463. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Fu, Y.; Xu, J.; Gong, G.-T.; Dai, J.-J.; Yi, J.; Liu, L. Pd(II)-Catalyzed C–H Activation/Aryl-Aryl Coupling of Phenol Esters. J. Am. Chem. Soc. 2010, 132, 468–469. [Google Scholar] [CrossRef]
- Ueta, Y.; Mikami, K.; Ito, S. Accessto Air-STable 1,3-Diphosphacyclobutane-2,4-Diyls by an Arylation Reaction with Arynes. Angew. Chem. Int. Ed. 2016, 55, 7525–7529. [Google Scholar] [CrossRef]
- Buzard, D.J.; Olsson, C.; Noson, K.; Lipshutz, B.H. A Modular Route to Nonracemic Cyclo-Nobins. Preparation of the Parent Ligand for Homo- and Heterogeneous Catalysis. Tetrahedron 2004, 60, 4443–4449. [Google Scholar]
- Schreiner, J.L.; Pirkle, W.H. Chiral High-Pressure Liquid Chromatographic Stationary Phases. Separation of the Enantiomers of Bi-β-naphthols and Analogues. J. Org. Chem. 1981, 46, 4988–4991. [Google Scholar]
- Kingsbury, W.D. Synthesis of Structural Analogs of Leukotriene B4 and Their Receptor Binding Activity. J. Med. Chem. 1993, 36, 3308–3320. [Google Scholar] [CrossRef]
- Wolsey, W.C. Perchlorate Salts, Their Uses and Alternatives. J. Chem. Educ. 1973, 50, A335–A337. [Google Scholar] [CrossRef]
- Theveau, L.; Bellini, R.; Dydio, P.; Szabo, Z.; Werf, A.; Sander, R.A.; Reek, J.N.K.; Moberg, C. Cofactor-Controlled Chirality of Tropoisomeric Ligand. Organometallics 2016, 35, 1956–1963. [Google Scholar] [CrossRef]
- Ahmed, I.; Clark, D.-A. Rapid Synthesis of 3,3′ Bis-Arylated BINOL Derivatives Using a C−H Borylation in Situ Suzuki−Miyaura Coupling Sequence. Org. Lett. 2014, 16, 4332–4335. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-J.; Clarkson, G.C.; Docherty, G.; North, C.L.; Woodward, G.; Wills, M. Ruthenium (II) Complexes of Monodonor Ligands: Efficient Reagents for Asymmetric Ketone Hydrogenation. J. Org. Chem. 2005, 70, 8079–8087. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Sun, X.; Li, X.-W.; Yuan, Y.-Z.; Jiao, N. Efficient and Practical Oxidative Bromination and Iodination of Arenes and Heteroarenes with DMSO and Hydrogen Halide: A Mild Protocol for Late-Stage Functionalization. Org. Lett. 2015, 17, 2886–2889. [Google Scholar] [CrossRef]
- Meesala, Y.; Wu, H.-L.; Koteswararao, B.; Kuo, T.-S.; Lee, W.-Z. Aerobic Oxidative Coupling of 2-Naphthol Derivatives Catalyzed by a Hexanuclear Bis(μ-hydroxo) Copper(II) Catalyst. Organometallics 2014, 33, 4385–4393. [Google Scholar] [CrossRef]
- Simonsen, K.B.; Gothelf, K.V.; Jørgensen, K.A. A Simple Synthetic Approach to 3,3′-Diaryl BINOLs. J. Org. Chem. 1998, 63, 7536–7538. [Google Scholar] [CrossRef]
- Qu, B.; Haddad, N. Ligand-Accelerated Stereoretentive Suzuki−Miyaura Coupling of Unprotected 3,3′-Dibromo-BINOL. J. Org. Chem. 2016, 81, 745–750. [Google Scholar] [CrossRef]
Sample Availability: Not available. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
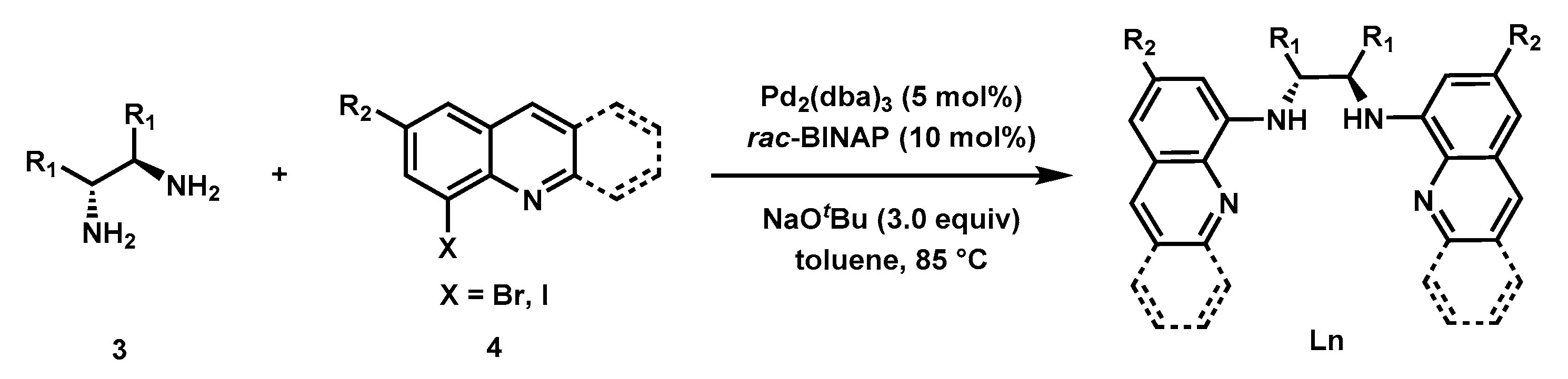{kind=link}
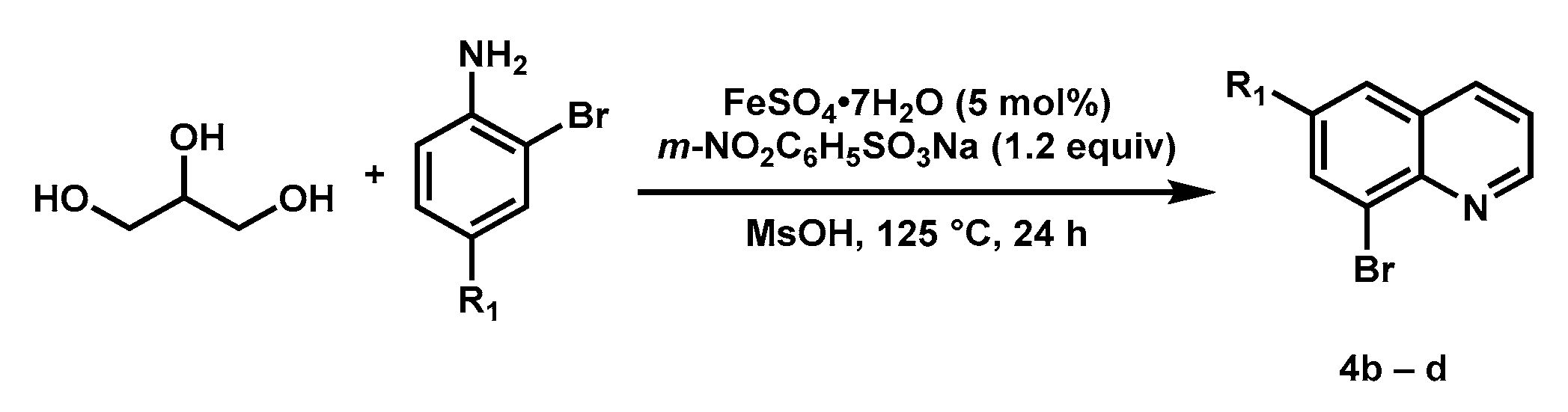{kind=link}
{kind=link}
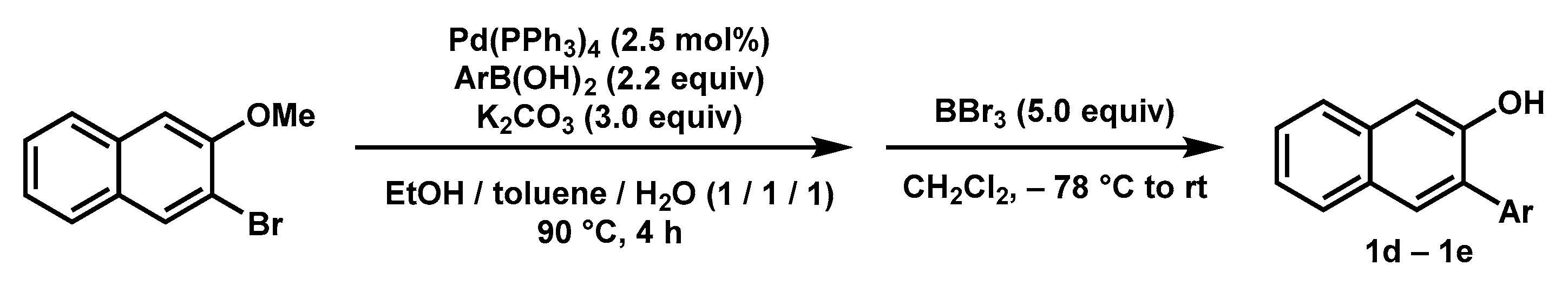{kind=link}

| Entry | Fe (x mol%) | Ln (y mol%) | Additives | Solvent | T °C | t/h | conv. (%) b | er (%) c |
|---|---|---|---|---|---|---|---|---|
| 1 | Fe(ClO4)2 (5.0) | L1(5.0) | - | MeOH | 30 | 28.5 | - | n.d. f |
| 2 | Fe(ClO4)2 (5.0) | L1 (5.0) | - | DCE | 30 | 28.5 | 57 d | 73:27 |
| 3 | Fe(ClO4)2 (5.0) | L1 (5.0) | - | CHCl3 | 30 | 28.5 | 68 d | 77:23 |
| 4 | Fe(ClO4)2 (5.0) | L1 (5.0) | - | toluene | 30 | 28.5 | 22 d | 80:20 |
| 5 | Fe(ClO4)2 (5.0) | L1 (5.0) | - | PhCl | 30 | 28.5 | 53 d | 79:21 |
| 6 | Fe(ClO4)2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 30 | 5.0 | 12 d | 80:20 |
| 7 | Fe(ClO4)2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 39 d | 78:22 |
| 8 | Fe(ClO4)2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 70 | 5.0 | 38 d | 73:27 |
| 9 | Fe(ClO4)2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 90 | 5.0 | 28 d | 70:30 |
| 10 | Fe(ClO4)3 (5.0) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 76 | 76:24 |
| 11 | Fe(OAc)2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 16 | n.d. f |
| 12 | Fe(OTf)2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 19 | n.d. f |
| 13 | Fe(acac)2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 23 | n.d. f |
| 14 | FeCl2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 20 | n.d. f |
| 15 | Fe(ClO4)2 (7.5) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 78 | 77:23 |
| 16 | Fe(ClO4)2 (10.0) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 88(84 e) | 80:20 |
| 17 | Fe(ClO4)2 (12.5) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 95 | 72:28 |
| 18 | Fe(ClO4)2 (5.0) | L1 (10.0) | MS 4Å | PhCl | 50 | 5.0 | 39 | 77:23 |
| 19 | Fe(ClO4)2 (10.0) | L1 (10.0) | MS 4Å | PhCl | 50 | 5.0 | 86 | 77:23 |
| 20 | Fe(ClO4)2 (10.0) | L2 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 90 | 70:30 |
| 21 | Fe(ClO4)2 (10.0) | L3 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 85 | 60:40 |
| 22 | Fe(ClO4)2 (10.0) | L4 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 53 e | 78:22 |
| 23 | Fe(ClO4)2 (10.0) | L5 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 67 e | 55:45 |
| 24 | Fe(ClO4)2 (10.0) | L6 (5.0) | MS 4Å | PhCl | 50 | 5.0 | - | n.d. f |
| 25 | Fe(ClO4)2 (10.0) | L7 (5.0) | MS 4Å | PhCl | 50 | 5.0 | - | n.d. f |
| 26 | Fe(ClO4)2 (10.0) | L8 (5.0) | MS 4Å | PhCl | 50 | 5.0 | - | n.d. f |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, L.-Y.; Usman, M.; Liu, W.-B. Enantioselective Iron/Bisquinolyldiamine Ligand-Catalyzed Oxidative Coupling Reaction of 2-Naphthols. Molecules 2020, 25, 852. https://doi.org/10.3390/molecules25040852
Wu L-Y, Usman M, Liu W-B. Enantioselective Iron/Bisquinolyldiamine Ligand-Catalyzed Oxidative Coupling Reaction of 2-Naphthols. Molecules. 2020; 25(4):852. https://doi.org/10.3390/molecules25040852
Chicago/Turabian StyleWu, Lin-Yang, Muhammad Usman, and Wen-Bo Liu. 2020. "Enantioselective Iron/Bisquinolyldiamine Ligand-Catalyzed Oxidative Coupling Reaction of 2-Naphthols" Molecules 25, no. 4: 852. https://doi.org/10.3390/molecules25040852
APA StyleWu, L.-Y., Usman, M., & Liu, W.-B. (2020). Enantioselective Iron/Bisquinolyldiamine Ligand-Catalyzed Oxidative Coupling Reaction of 2-Naphthols. Molecules, 25(4), 852. https://doi.org/10.3390/molecules25040852