Efficient Biocatalytic Synthesis of Dihalogenated Purine Nucleoside Analogues Applying Thermodynamic Calculations

 , , , , ,
, , , , ,  , and
, and
Abstract
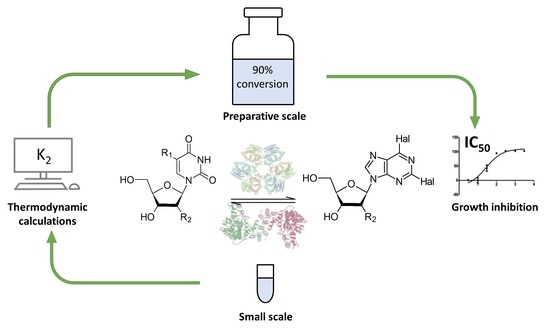
:
1. Introduction
2. Results
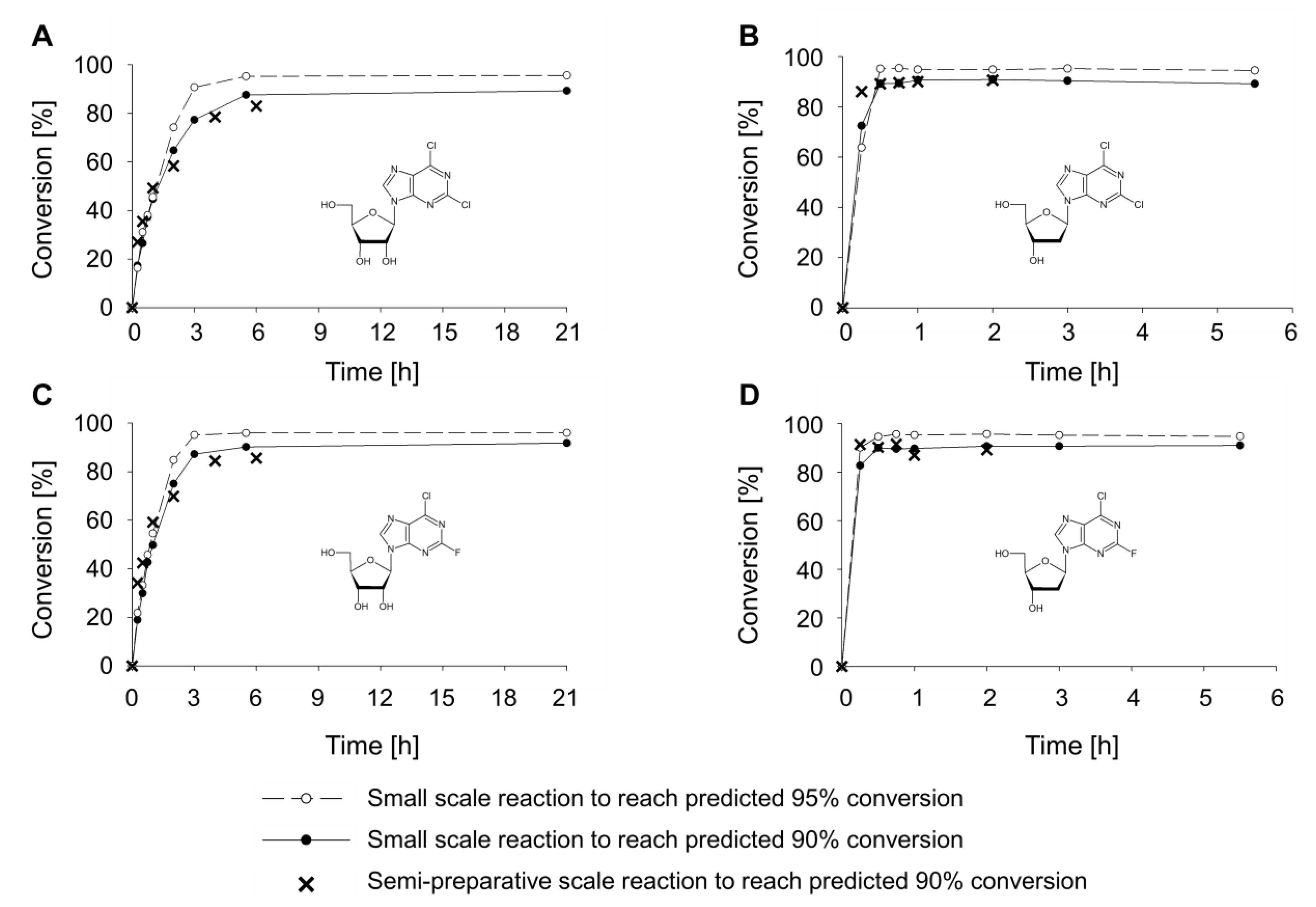
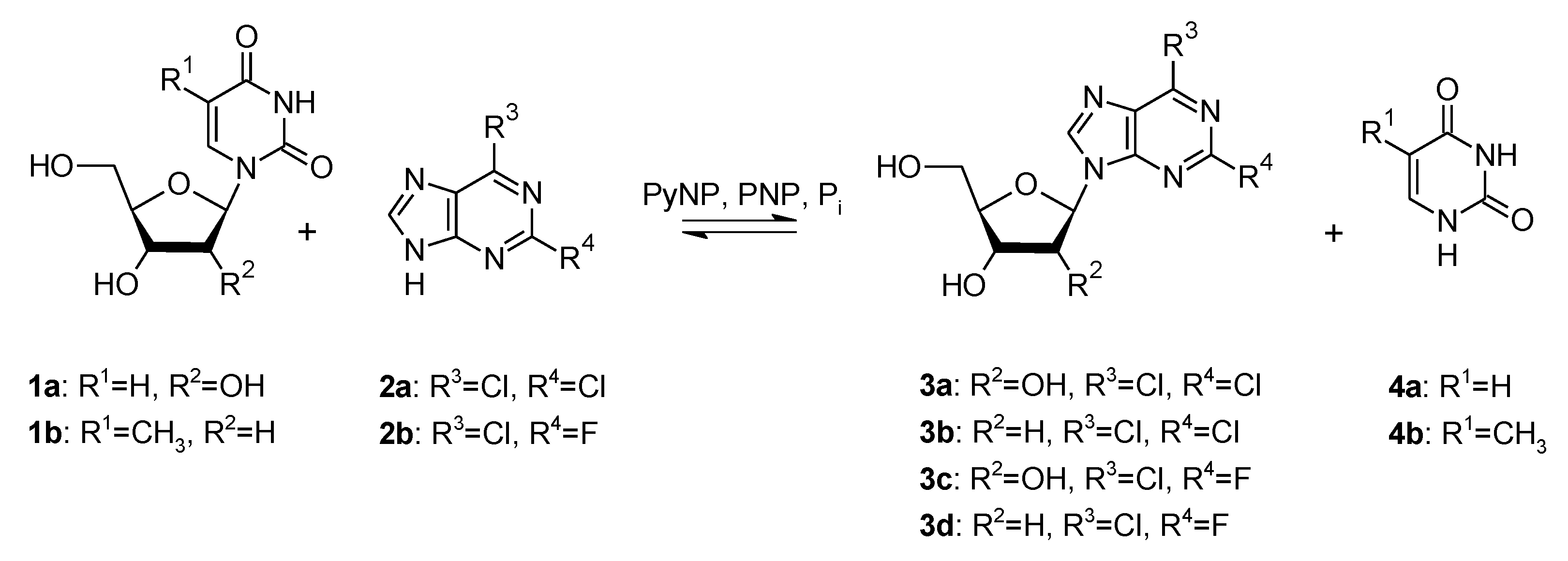
2.1. Optimization of the Synthesis of Dihalogenated Nucleosides Based on Thermodynamic Calculations
2.2. Synthesis of Dihalogenated Nucleosides in Semi-Preparative Scale
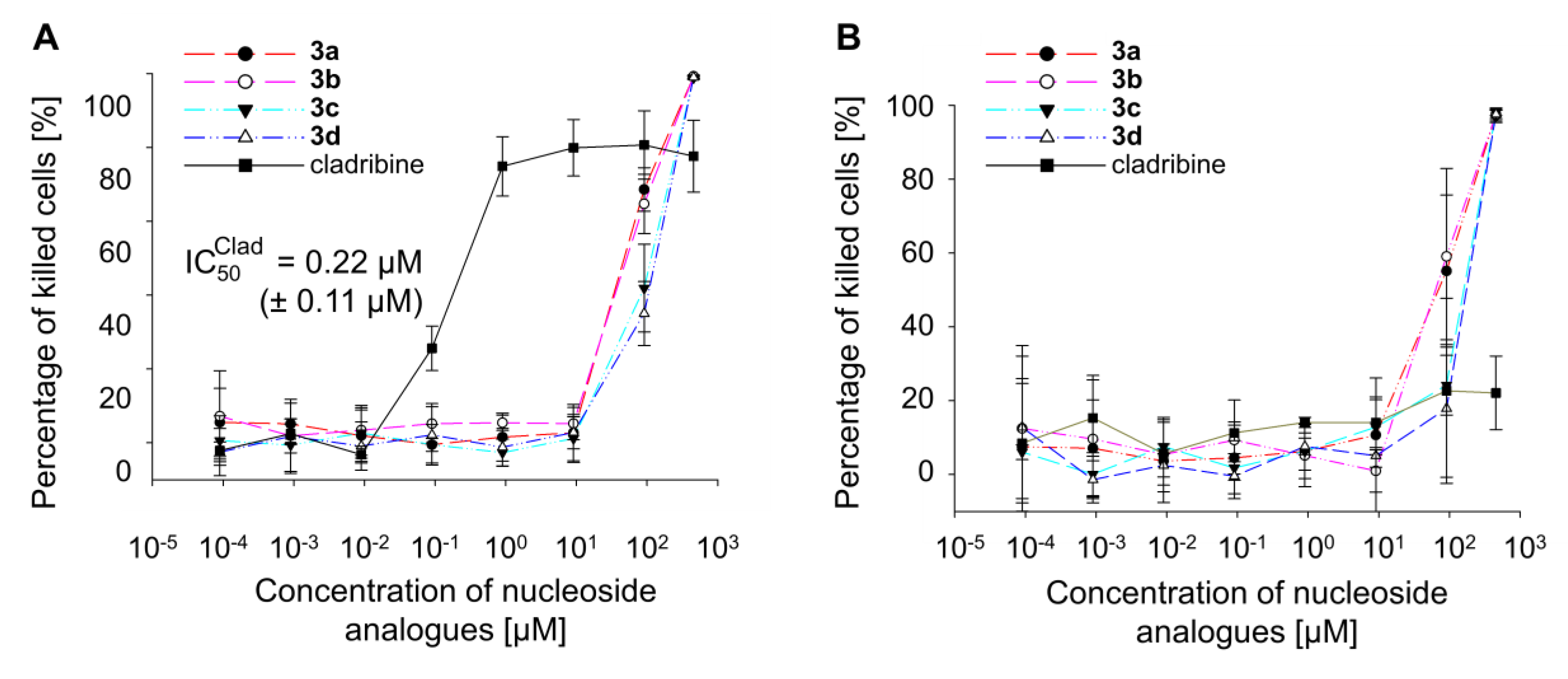
2.3. Effect of Dihalogenated Nucleoside Analogues on Cell Growth
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Optimization of the Enzymatic Synthesis of Dihalogenated Purine Nucleosides
4.3. Enzymatic Production of Dihalogenated Purine Nucleosides
4.4. Cell Culture
4.5. XTT Cell Proliferation Assay
4.6. Analytical HPLC
4.7. Purification of Nucleoside Analogues by Semi-Preparative HPLC
4.8. NMR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Trelles, J.A.; Bentancor, L.; Schoijet, A.; Porro, S.; Lewkowicz, E.S.; Sinisterra, J.V.; Iribarren, A.M. Immobilized Escherichia Coli BL21 as a Catalyst for the Synthesis of Adenine and Hypoxanthine Nucleosides. Chem. Biodivers. 2004, 1, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Rocchietti, S.; Ubiali, D.; Terreni, M.; Albertini, A.M.; Fernandez-Lafuente, R.; Guisan, J.M.; Pregnolato, M. Immobilization and Stabilization of Recombinant Multimeric Uridine and Purine Nucleoside Phosphorylases from Bacillus Subtilis. Biomacromolecules 2004, 5, 2195–2200. [Google Scholar] [CrossRef] [PubMed]
- Rivero, C.W.; Britos, C.N.; Lozano, M.E.; Sinisterra, J.V.; Trelles, J.A. Green Biosynthesis of Floxuridine by Immobilized Microorganisms. FEMS Microbiol. Lett. 2012, 331, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iglesias, L.E.; Lewkowicz, E.S.; Medici, R.; Bianchi, P.; Iribarren, A.M. Biocatalytic Approaches Applied to the Synthesis of Nucleoside Prodrugs. Biotechnol. Adv. 2015, 33, 412–434. [Google Scholar] [CrossRef]
- Haki, G.D.; Rakshit, S.K. Developments in Industrially Important Thermostable Enzymes: A Review. Bioresour. Technol. 2003, 89, 17–34. [Google Scholar] [CrossRef]
- Bruins, M.; Janssen, A.; Boom, R. Thermozymes and Their Applications. Appl. Biochem. Biotechnol. 2001, 90, 155–186. [Google Scholar] [CrossRef]
- Quan, D.J.; Peters, M.G. Antiviral Therapy: Nucleotide and Nucleoside Analogs. Clin. Liver Dis. 2004, 8, 371–385. [Google Scholar] [CrossRef]
- Robak, P.; Robak, T. Older and New Purine Nucleoside Analogs for Patients with Acute Leukemias. Cancer Treat. Rev. 2013, 39, 851–861. [Google Scholar] [CrossRef]
- Khaw, M.; Panosian, C.B. Human Antiprotozoal Therapy: Past, Present, and Future. Clin. Microbiol. Rev. 1995, 8, 427–439. [Google Scholar] [CrossRef]
- Bennett, L., Jr.; Chang, C.-H.; Allan, P.W.; Adamson, D.J.; Rose, L.M.; Brockman, R.W.; Secrist, J.A., III; Shortnacy, A.; Montgomery, J.A. Metabolism and Metabolic Effects of Halopurine Nucleosides in Tumor Cells in Culture. Nucleosides Nucleotides 1985, 4, 107–116. [Google Scholar] [CrossRef]
- Huang, M.-C.; Hatfield, K.; Roetker, A.W.; Montgomery, J.A.; Blakley, R.L. Analogs of 2′-Deoxyadenosine: Facile Enzymatic Preparation and Growth Inhibitory Effects on Human Cell Lines. Biochem. Pharmacol. 1981, 30, 2663–2671. [Google Scholar] [CrossRef]
- Huang, M.C.; Montgomery, J.A.; Thorpe, M.C.; Stewart, E.L.; Secrist, J.A.; Blakley, R.L. Formation of 3-(2′-Deoxyribofuranosyl) and 9-(2′-Deoxyribofuranosyl) Nucleosides of 8-Substituted Purines by Nucleoside Deoxyribosyltransferase. Arch. Biochem. Biophys. 1983, 222, 133–144. [Google Scholar] [CrossRef]
- Kaspar, F.; Giessmann, R.T.; Hellendahl, K.; Neubauer, P.; Wagner, A.; Gimpel, M. General Principles for Yield Optimization of Nucleoside Phosphorylase-catalyzed Transglycosylations. ChemBioChem 2019. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, F.; Giessmann, R.T.; Neubauer, P.; Wagner, A.; Gimpel, M. Thermodynamic Reaction Control of Nucleoside Phosphorolysis. Adv. Synth. Catal. 2020. [Google Scholar] [CrossRef] [Green Version]
- Kaspar, F.; Giessman, R.T. Supplementary Material for “General Principles for Yield Optimization of Nucleoside Phosphorylase-Catalyzed Transglycosylations”. Zenodo 2019. [Google Scholar] [CrossRef]
- Giessmann, R.T. A Python Toolbox to Evaluate Systems under Thermodynamic Reaction Control. Zenodo 2019. [CrossRef]
- Kaspar, F.; Wagner, A. Supplementary Material for “Efficient Biocatalytic Synthesis of Dihalogenated Purine Nucleoside Analogues Applying Thermodynamic Calculations”. Zenodo 2020. [Google Scholar] [CrossRef]
- Stachnik, K.; Grieb, P.; Skierski, J.S. Cytotoxic Effects of Cladribine and Tezacitabine toward HL-60. Acta Biochim. Pol. 2005, 52, 561–565. [Google Scholar] [CrossRef]
- Nishi, R.; Yamauchi, T.; Negoro, E.; Takemura, H.; Ueda, T. Combination of Guanine Arabinoside and Bcl-2 Inhibitor YC137 Overcomes the Cytarabine Resistance in HL-60 Leukemia Cell Line. Cancer Sci. 2013, 104, 502–507. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Szeker, K.; Jiao, L.-Y.; Oestreich, M.; Mikhailopulo, I.A.; Neubauer, P. Synthesis of 2,6-Dihalogenated Purine Nucleosides by Thermostable Nucleoside Phosphorylases. Adv. Synth. Catal. 2015, 357, 1237–1244. [Google Scholar] [CrossRef]
- Zhou, X.; Mikhailopulo, I.A.; Cruz Bournazou, M.N.; Neubauer, P. Immobilization of Thermostable Nucleoside Phosphorylases on MagReSyn® Epoxide Microspheres and Their Application for the Synthesis of 2,6-Dihalogenated Purine Nucleosides. J. Mol. Catal. B Enzym. 2015, 115, 119–127. [Google Scholar] [CrossRef]
- Alexeev, C.S.; Kulikova, I.V.; Gavryushov, S.; Tararov, V.I.; Mikhailov, S.N. Quantitative Prediction of Yield in Transglycosylation Reaction Catalyzed by Nucleoside Phosphorylases. Adv. Synth. Catal. 2018, 360, 3090–3096. [Google Scholar] [CrossRef]
- Faderl, S.; Gandhi, V.; Keating, M.J.; Jeha, S.; Plunkett, W.; Kantarjian, H.M. The Role of Clofarabine in Hematologic and Solid Malignancies—Development of a next-Generation Nucleoside Analog. Cancer 2005, 103, 1985–1995. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-C.; Luo, M.-Z.; Mozdziesz, D.E.; Sartorelli, A.C. Synthesis and Biological Evaluation of 2- and 7-Substituted 9-Deazaadenosine Analogues. Nucleosides Nucleotides Nucleic Acids 2005, 24, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, S.; Zhao, M.; Deng, X.-S.; Lee, C.-K.; Yu, X.-D.; Liu, B. Therapeutic Potential of Cladribine in Combination with STAT3 Inhibitor against Multiple Myeloma. BMC Cancer 2011, 11, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pui, C.-H.; Jeha, S.; Kirkpatrick, P. Clofarabine. Nat. Rev. Drug. Discov. 2005, 4, 369–370. [Google Scholar] [CrossRef]
- Pastor-Anglada, M.; Molina-Arcas, M.; Casado, F.J.; Bellosillo, B.; Colomer, D.; Gil, J. Nucleoside Transporters in Chronic Lymphocytic Leukaemia. Leukemia 2004, 18, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Deville-Bonne, D.; El Amri, C.; Meyer, P.; Chen, Y.; Agrofoglio, L.A.; Janin, J. Human and Viral Nucleoside/Nucleotide Kinases Involved in Antiviral Drug Activation: Structural and Catalytic Properties. Antiviral. Res. 2010, 86, 101–120. [Google Scholar] [CrossRef]
- Freyer, C.W.; Gupta, N.; Wetzler, M.; Wang, E.S. Revisiting the Role of Cladribine in Acute Myeloid Leukemia: An Improvement on Past Accomplishments or More Old News? Am. J. Hematol. 2015, 90, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Warzocha, K.; Fabianowska-Majewska, K.; Bloński, J.; Krykowski, E.; Robak, T. 2-Chlorodeoxyadenosine Inhibits Activity of Adenosine Deaminase and S-Adenosylhomocysteine Hydrolase in Patients with Chronic Lymphocytic Leukaemia. Eur. J. Cancer 1997, 33, 170–173. [Google Scholar] [CrossRef]
- Carrera, C.J.; Terai, C.; Lotz, M.; Curd, J.G.; Piro, L.D.; Beutler, E.; Carson, D.A. Potent Toxicity of 2-Chlorodeoxyadenosine toward Human Monocytes in Vitro and in Vivo: A Novel Approach to Immunosuppressive Therapy. J. Clin. Investig. 1990, 86, 1480–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorbrüggen, H.; Ruh-Pohlenz, C. Synthesis of Nucleosides. In Organic Reactions; John Wiley & Sons: Hoboken, NJ, USA, 2004; pp. 1–630. [Google Scholar]
- Chemo-Enzymatic Preparation Method for Purine Nucleosides and Their Deaza- and Aza-analogues. Available online: https://patents.google.com/patent/WO2016034735A1/en (accessed on 22 January 2020).
- Kaspar, F.; Giessmann, R.T.; Krausch, N.; Neubauer, P.; Wagner, A.; Gimpel, M. A UV/Vis Spectroscopy-Based Assay for Monitoring of Transformations between Nucleosides and Nucleobases. Methods Protoc. 2019, 2, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, J.; Hiller, T.; Kissner, M.S.; Qazi, T.H.; Duda, G.N.; Hocke, A.C.; Hippenstiel, S.; Elomaa, L.; Weinhart, M.; Fahrenson, C.; et al. Optimization of Cell-Laden Bioinks for 3D Bioprinting and Efficient Infection with Influenza A Virus. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Szeker, K.; Janocha, B.; Böhme, T.; Albrecht, D.; Mikhailopulo, I.A.; Neubauer, P. Recombinant Purine Nucleoside Phosphorylases from Thermophiles: Preparation, Properties and Activity towards Purine and Pyrimidine Nucleosides. FEBS J. 2013, 280, 1475–1490. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.K.; Becker, E.D.; Cabral De Menezes, S.M.; Goodfellow, R.; Granger, P. NMR Nomenclature. Nuclear Spin Properties and Conventions for Chemical Shifts (IUPAC Recommendations 2001). Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds DCR-R, 3b, 3c and 3d are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product | Product Formation [%] at Equilibrium * | Equilibrium Constant K2 ** | Sugar Donor Excess to Reach: | |
|---|---|---|---|---|
| 90% Product Yield | 95% Product Yield | |||
| 3a | 60 | 0.076 | 4.5 | 9.0 |
| 3b | 55 | 0.081 | 6.1 | 12.5 |
| 3c | 60 | 0.076 | 4.4 | 8.8 |
| 3d | 57 | 0.071 | 5.4 | 11.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yehia, H.; Westarp, S.; Röhrs, V.; Kaspar, F.; Giessmann, R.T.; Klare, H.F.T.; Paulick, K.; Neubauer, P.; Kurreck, J.; Wagner, A. Efficient Biocatalytic Synthesis of Dihalogenated Purine Nucleoside Analogues Applying Thermodynamic Calculations. Molecules 2020, 25, 934. https://doi.org/10.3390/molecules25040934
Yehia H, Westarp S, Röhrs V, Kaspar F, Giessmann RT, Klare HFT, Paulick K, Neubauer P, Kurreck J, Wagner A. Efficient Biocatalytic Synthesis of Dihalogenated Purine Nucleoside Analogues Applying Thermodynamic Calculations. Molecules. 2020; 25(4):934. https://doi.org/10.3390/molecules25040934
Chicago/Turabian StyleYehia, Heba, Sarah Westarp, Viola Röhrs, Felix Kaspar, Robert T. Giessmann, Hendrik F.T. Klare, Katharina Paulick, Peter Neubauer, Jens Kurreck, and Anke Wagner. 2020. "Efficient Biocatalytic Synthesis of Dihalogenated Purine Nucleoside Analogues Applying Thermodynamic Calculations" Molecules 25, no. 4: 934. https://doi.org/10.3390/molecules25040934
APA StyleYehia, H., Westarp, S., Röhrs, V., Kaspar, F., Giessmann, R. T., Klare, H. F. T., Paulick, K., Neubauer, P., Kurreck, J., & Wagner, A. (2020). Efficient Biocatalytic Synthesis of Dihalogenated Purine Nucleoside Analogues Applying Thermodynamic Calculations. Molecules, 25(4), 934. https://doi.org/10.3390/molecules25040934