
Effect of Heat Treatment on the Chemical Structure and Thermal Properties of Softwood-Derived Glycol Lignin

 , , ,
, , ,
Abstract
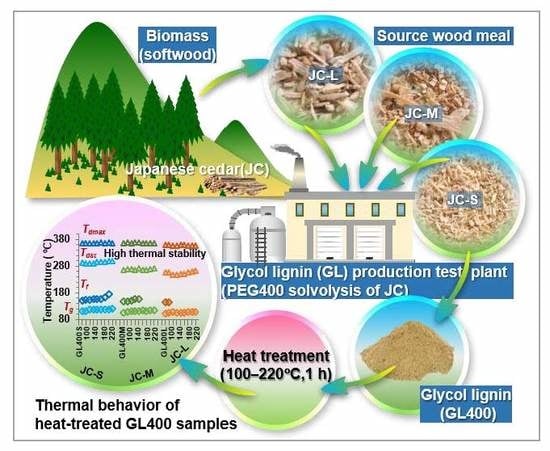
:
1. Introduction
2. Results and Discussion
2.1. Lignin Content of Heat-Treated GL400
2.2. Molecular Weight Distribution of Heat-Treated GL400
2.3. Chemical Structure of Heat-Treated GL400 Samples
2.4. Thermal Properties of Heat-Treated GL400 Samples
3. Materials and Methods
3.1. GL Preparation and Heat Treatment
3.2. Chemical Analyses
3.3. D NMR
3.4. ATR-FTIR Spectroscopy
3.5. SEC
3.6. TMA
3.7. TGA
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Henriksson, G. Lignin. In Pulp and Paper Chemistry and Technology, Wood Chemistry and Wood Biotechnology; Ek, M., Gellerstedt, G., Henriksson, G., Eds.; de Grutyter: Berlin, Germany, 2009; Volume 1, pp. 121–146. [Google Scholar]
- Brännvall, E. Overview of Pulp and Paper Processes. In Pulp and Paper Chemistry and Technology, Pulping Chemistry and Technology; Ek, M., Gellerstedt, G., Henrikkson, G., Eds.; de Grutyter: Berlin, Germany, 2009; Volume 2, pp. 1–12. [Google Scholar]
- Chiang, V.L.; Puumala, R.J.; Takeuchi, H.; Eckert, R.E. Comparison of softwood and hardwood kraft pulping. Tappi. J. 1988, 71, 173–176. [Google Scholar]
- Lia, Y.Z.; Sarkanen, K.V. Isolation and Structural Studies. In Lignins, Occurrence, Formation, Structure and Reactions; Sarkanen, K.V., Ludwig, C.H., Eds.; Wiley Interscience: New York, NY, USA, 1971; pp. 165–240. [Google Scholar]
- Brunow, G.; Lundquist, K. Functional Groups and Bonding Patterns in Lignin (Including the Lignin-Carbohydrate Complexes). In Lignin and Lignans: Advances in Chemistry; Heitner, C., Dimmel, D., Schmidt, J., Eds.; CRC Press: Boca Rotan, FL, USA, 2010; pp. 267–299. [Google Scholar]
- Norgren, M.; Edlund, H. Lignin: Recent advances and emerging applications. Curr. Opin. in Colloid In. 2014, 19, 409–416. [Google Scholar] [CrossRef]
- Li, J.; Henriksson, G.; Gellerstedt, G. Lignin depolymerization/repolymerization and its critical role for delignification of aspen wood by steam explosion. Bioresour. Technol. 2007, 98, 3061–3068. [Google Scholar] [CrossRef]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin biosynthesis and structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Öhman, F.; Theliander, H.; Tomani, P.; Axegard, P. Method for Separating Lignin from Black Liquor. U.S. Patent 8486224B2, 16 July 2013. [Google Scholar]
- Kouisni, L.; Paleologou, M. Method for Separating Lignin from Black Liquor. U.S. Patent 8771464B2, 8 July 2014. [Google Scholar]
- Lange, H.; Schiffels, P.; Sette, M.; Sevastyanova, O.; Crestini, C. Fractional precipitation of wheat straw organosolv lignin: Macroscopic properties and structural insights. ACS Sustainable Chem. Eng. 2016, 4, 5136–5151. [Google Scholar] [CrossRef]
- Nitsos, C.; Stoklosa, R.; Karnaouri, A.; Vörös, D.; Lange, H.; Hodge, D.; Crestini, C.; Rova, U.; Christakopoulos, P. Isolation and characterization of organosolv and alkaline lignins from hardwood and softwood biomass. ACS Sustainable Chem. Eng. 2016, 4, 5181–5193. [Google Scholar] [CrossRef]
- Jiang, X.; Savithri, D.; Du, X.; Pawar, S.; Jameel, H.; Chang, H.-M.; Zhou, X. Fractionation and characterization of kraft lignin by sequential precipitation with various organic solvent. ACS Sustainable Chem. Eng. 2017, 5, 835–842. [Google Scholar] [CrossRef]
- Yawalata, D.; Paszner, L. Anionic effect in high concentration alcohol organosolv pulping. Holzforschung 2004, 58, 1–6. [Google Scholar] [CrossRef]
- Dence, C.W.; Lin, S.Y. Introduction. In Methods in Lignin Chemistry: Springer Series in Wood Science; Dence, C.W., Lin, S.Y., Eds.; Springer: Berlin, Germany, 1992; pp. 1–19. [Google Scholar]
- Nge, T.T.; Tobimatsu, Y.; Takahashi, S.; Takata, E.; Yamamura, M.; Miyagawa, Y.; Ikeda, T.; Umezawa, T.; Yamada, T. Isolation and characterization of polyethylene glycol (PEG)-modified glycol lignin via PEG solvolysis of softwood biomass in a large-scale batch reactor. ACS Sustainable Chem. Eng. 2018, 6, 7841–7848. [Google Scholar] [CrossRef]
- Nge, T.T.; Takata, E.; Takahashi, S.; Yamada, T. Isolation and thermal characterization of softwood-derived lignin with thermal flow properties. ACS Sustainable Chem. Eng. 2016, 4, 2861–2868. [Google Scholar] [CrossRef]
- Kaneko, H.; Ishii, R.; Suzuki, A.; Nakamura, T.; Ebina, T.; Nge, T.T.; Yamada, T. Flexible clay glycol lignin nanocomposite film with heat durability and high moisture-barrier property. Appl. Clay Sci. 2016, 132–133, 425–429. [Google Scholar] [CrossRef]
- Takahashi, K.; Ishii, R.; Nakamura, T.; Suzuki, A.; Ebina, T.; Yoshida, M.; Kubota, M.; Nge, T.T.; Yamada, T. Flexible electronic substrate film fabricated using natural clay and wood components with cross-linking polymer. Adv. Mater. 2017, 29, 1606512. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kobayashi, F.; Ebina, T.; Ishii, R.; Nakamura, T.; Nge, T.T.; Yamada, T.; Shiraishi, A.; Yamashita, T. Effect of thermal base generators on the FRP fabrication with glycol-lignin. J. Photopolym. Sci. Tec. 2018, 31, 101–106. [Google Scholar] [CrossRef]
- Ono, K.; Tanaike, O.; Ishii, R.; Nakamura, T.; Shikinaka, K.; Ebina, T.; Nge, T.T.; Yamada, T. Solvent-free fabrication of an elastomeric epoxy resin using glycol lignin from Japanese cedar. ACS Omega 2019, 4, 17251–17256. [Google Scholar] [CrossRef]
- Suzuki, A.; Shikinaka, K.; Ishii, R.; Yoshida, H.; Ebina, T.; Ishida, T.; Nge, T.T.; Yamada, T. Heat-resistant insulation film containing clay and wood components. Appl. Clay Sci. 2019, 180, 105189. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Hwang, H.; Oh, S.; Kim, Y.-S.; Kim, U.-J.; Choi, J.W. Investigation of structural modification and thermal characteristics of lignin after heat treatment. Int. J. Biol. Macromol. 2014, 66, 57–65. [Google Scholar] [CrossRef]
- Rousset, P.; Lapierre, C.; Pollet, B.; Quirino, W.; Perre, P. Effect of severe thermal treatment on spruce and beech wood lignins. Ann. For. Sci. 2009, 66, 110. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, H.; Sun, S.; Cao, X.; Sun, R. Effect of hydrothermal pretreatment on the structural changes of alkali ethanol lignin from wheat straw. Sci. Rep. 2016, 6, 39354. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, J.-Q.; Li, H.; Yuan, T.-Q.; Wang, Y.-Y.; Sun, R.-C. Heat treatment of industrial alkaline lignin and its potential application as an adhesive for green wood-lignin composites. ACS Sustainable Chem. Eng. 2017, 5, 7269–7277. [Google Scholar] [CrossRef]
- Matsuura, H.; Miyazawa, T. Interchain force field and normal vibrations of polyethylene glycol. Bull. Chem. Soc. Jpn. 1968, 41, 1798–1808. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Lan, J.; Ma, P.; Dong, X.; Qu, J.; He, H. Chemical structure and thermal properties of lignin modified with polyethylene glycol during steam explosion. Wood Sci Technol. 2017, 51, 135–150. [Google Scholar] [CrossRef]
- Faix, O. Fourier Transform Infrared Spectroscopy. In Methods in Lignin Chemistry: Springer Series in Wood Science; Dence, C.W., Lin, S.Y., Eds.; Springer: Berlin, Germany, 1992; pp. 83–110. [Google Scholar]
- Sun, Q.; Khunsupat, R.; Akato, K.; Tao, J.; Labbé, N.; Gallego, N.C.; Bozell, J.J.; Rials, T.G.; Tuskan, G.A.; Tschaplinski, T.J.; et al. A study of poplar organosolv lignin after melt rheology treatment as carbon fiber precursors. Green Chem. 2016, 18, 5015–5024. [Google Scholar] [CrossRef]
- Yamamura, M.; Hattori, T.; Suzuki, S.; Shibata, D.; Umezawa, T. Microscale thioacidolysis method for the rapid analysis of β–O–4 substructures in lignin. Plant Biotechnol. 2012, 29, 419–423. [Google Scholar] [CrossRef] [Green Version]
- Lapierre, C.; Monties, B.; Rolando, C. Preparative thioacidolysis of spruce lignin: Isolation and identification of main monomeric products. Holzforschung 1986, 40, 47–50. [Google Scholar] [CrossRef]
- Sen, S.; Patil, S.; Argyropoulos, D.S. Thermal properties of lignin in copolymers, blends, and composites: A Review. Green Chem. 2015, 17, 4862–4887. [Google Scholar] [CrossRef]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D.; Crocker, D. Determination of Structural Carbohydrates and Lignin in Biomass; National Renewable Energy Laboratory: Golden, CO, USA, 2008.
- Yue, F.; Lu, F.; Sun, R.-C.; Ralph, J. Syntheses of lignin-derived thioacidolysis monomers and their uses as quantitation standards. J. Agric. Food. Chem. 2012, 60, 922–928. [Google Scholar] [CrossRef]
- Lam, P.Y.; Tobimatsu, Y.; Takeda, Y.; Suzuki, S.; Yamamura, M.; Umezawa, T.; Lo, C. Disruption flavone synthase II alters lignin and improves biomass digestibility. Plant Physiol. 2017, 174, 972–985. [Google Scholar] [CrossRef] [Green Version]
- Mansfield, S.D.; Kim, H.; Lu, F.; Ralph, J. Whole plant cell wall characterization using solution-state 2D NMR. Nat. Protoc. 2012, 7, 1579–1589. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thioacidolysis Monomer Yield (μmol/g) b | Relative NMR Signal intensity and Ratio c | Molecular Mass d | |||||||
| GL Samples a | Klason Lignin (wt%) | per GL | per Klason Lignin | I’ [β–O–4] (%) | II[β–5] (%) | III[β–β] (%) | P2/OMeRatio | Mw | Mn |
| GL400S | 79.8 | 14.7 | 18.4 | 32 | 20 | 49 | 0.30 | 5310 | 2050 |
| GL400S-100 | - | 15.6 | - | 35 | 22 | 43 | 0.30 | 5450 | 2060 |
| GL400S-120 | - | 15.2 | - | - | - | - | - | 6150 | 2130 |
| GL400S-140 | - | 15.9 | - | - | - | - | - | 6910 | 2150 |
| GL400S-150 | - | 15.8 | - | - | - | - | - | 7650 | 2220 |
| GL400S-160 | 80.7 | 13.8 | 17.0 | 23 | 24 | 52 | 0.29 | 9220 | 2280 |
| GL400S-180 | 80.8 | 16.2 | 20.0 | - | - | - | - | 17,900 * | 2530 * |
| GL400S-200 | 81.3 | 13.7 | 16.9 | - | - | - | - | 48,840 * | 2380 * |
| GL400S-220 | 80.2 | 14.5 | 18.1 | 10 * | 23 * | 67 * | 0.32 * | 7060 * | 1720 * |
| GL400M | 78.0 | 81.8 | 104.9 | 50 | 31 | 19 | 0.30 | 8150 | 2360 |
| GL400M-100 | - | 82.0 | - | 52 | 31 | 16 | 0.29 | 8660 | 2400 |
| GL400M-120 | - | 81.7 | - | - | - | - | - | 10,830 | 2500 |
| GL400M-140 | - | 85.6 | - | - | - | - | - | 22,640 | 2710 |
| GL400M-150 | - | 81.3 | - | - | - | - | - | 21,770 * | 2490 |
| GL400M-160 | 79.4 | 81.8 | 103.1 | 48 | 32 | 20 | 0.27 | 10,090 * | 2000 * |
| GL400M-180 | 79.1 | 79.9 | 101.0 | - | - | - | - | 4260 * | 1470 * |
| GL400M-200 | 78.9 | 71.9 | 91.2 | - | - | - | - | 2840 * | 1210 * |
| GL400M-220 | 79.9 | 51.8 | 64.9 | 7 * | 47 * | 46 * | 0.62 * | 2110 * | 1040 * |
| GL400L | 75.1 | 168.7 | 224.7 | 52 | 36 | 12 | 0.35 | 13,630 | 2590 |
| GL400L-100 | - | 167.2 | - | 53 | 35 | 11 | 0.33 | 17,170 | 2680 |
| GL400L-120 | - | 161.8 | - | - | - | - | - | 16,180 * | 2380 * |
| GL400L-140 | - | 159.4 | - | - | - | - | - | 7090 * | 1910 * |
| GL400L-150 | - | 148.6 | - | - | - | - | - | 5730 * | 1760 * |
| GL400L-160 | 76.6 | 156.1 | 203.7 | 48 | 40 | 12 | 0.29 | 4930 * | 1630 * |
| GL400L-180 | 77.3 | 138.0 | 178.5 | - | - | - | - | 2920 * | 1240 * |
| GL400L-200 | 77.8 | 109.4 | 140.5 | - | - | - | - | 2260 * | 1080 * |
| GL400L-220 | 79.3 | 91.9 | 115.8 | 20 * | 60 * | 20 * | 0.89 * | 1840 * | 970 * |
| GL400L | Gl400L-200 | GL400L-220 | |||||||
| a Soluble and Insoluble | b Insoluble Fraction | a Soluble and Insoluble | b Insoluble Fraction | ||||||
| λ−1/cm−1 | c RA | λ−1/cm−1 | c RA | λ−1/cm−1 | c RA | λ−1/cm−1 | c RA | λ−1/cm−1 | c RA |
| d 947 | 0.265 | 949 | 0.270 | 949 | 0.223 | 949 | 0.271 | 951 | 0.245 |
| e 1350 | 0.483 | 1350 | 0.484 | 1354 | 0.628 | 1350 | 0.487 | 1356 | 0.645 |
| f 1423 | 0.521 | 1425 | 0.534 | 1421 | 0.673 | 1427 | 0.542 | 1421 | 0.735 |
| g 1462 | 0.730 | 1462 | 0.746 | 1460 | 0.890 | 1462 | 0.756 | 1460 | 0.952 |
| h 1597 | 0.448 | 1597 | 0.465 | 1593 | 0.702 | 1597 | 0.470 | 1593 | 0.709 |
| GL400M | Gl400M-200 | GL400M-220 | |||||||
| a Soluble and Insoluble | b Insoluble Fraction | a Soluble and Insoluble | b Insoluble Fraction | ||||||
| λ−1/cm−1 | c RA | λ−1/cm−1 | c RA | λ−1/cm−1 | c RA | λ−1/cm−1 | c RA | λ−1/cm−1 | c RA |
| d 949 | 0.223 | 949 | 0.258 | 949 | 0.244 | 949 | 0.280 | 949 | 0.240 |
| e 1350 | 0.472 | 1350 | 0.484 | 1354 | 0.597 | 1350 | 0.494 | 1354 | 0.636 |
| f 1427 | 0.508 | 1427 | 0.540 | 1423 | 0.645 | 1427 | 0.559 | 1421 | 0.684 |
| g 1462 | 0.725 | 1462 | 0.756 | 1460 | 0.874 | 1462 | 0.769 | 1460 | 0.919 |
| h 1597 | 0.431 | 1597 | 0.460 | 1593 | 0.679 | 1597 | 0.468 | 1593 | 0.709 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nge, T.T.; Tobimatsu, Y.; Yamamura, M.; Takahashi, S.; Takata, E.; Umezawa, T.; Yamada, T. Effect of Heat Treatment on the Chemical Structure and Thermal Properties of Softwood-Derived Glycol Lignin. Molecules 2020, 25, 1167. https://doi.org/10.3390/molecules25051167
Nge TT, Tobimatsu Y, Yamamura M, Takahashi S, Takata E, Umezawa T, Yamada T. Effect of Heat Treatment on the Chemical Structure and Thermal Properties of Softwood-Derived Glycol Lignin. Molecules. 2020; 25(5):1167. https://doi.org/10.3390/molecules25051167
Chicago/Turabian StyleNge, Thi Thi, Yuki Tobimatsu, Masaomi Yamamura, Shiho Takahashi, Eri Takata, Toshiaki Umezawa, and Tatsuhiko Yamada. 2020. "Effect of Heat Treatment on the Chemical Structure and Thermal Properties of Softwood-Derived Glycol Lignin" Molecules 25, no. 5: 1167. https://doi.org/10.3390/molecules25051167
APA StyleNge, T. T., Tobimatsu, Y., Yamamura, M., Takahashi, S., Takata, E., Umezawa, T., & Yamada, T. (2020). Effect of Heat Treatment on the Chemical Structure and Thermal Properties of Softwood-Derived Glycol Lignin. Molecules, 25(5), 1167. https://doi.org/10.3390/molecules25051167