
A 1,5-Oligosilanylene Dianion as Building Block for Oligosiloxane Containing Cages, Ferrocenophanes, and Cyclic Germylenes and Stannylenes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
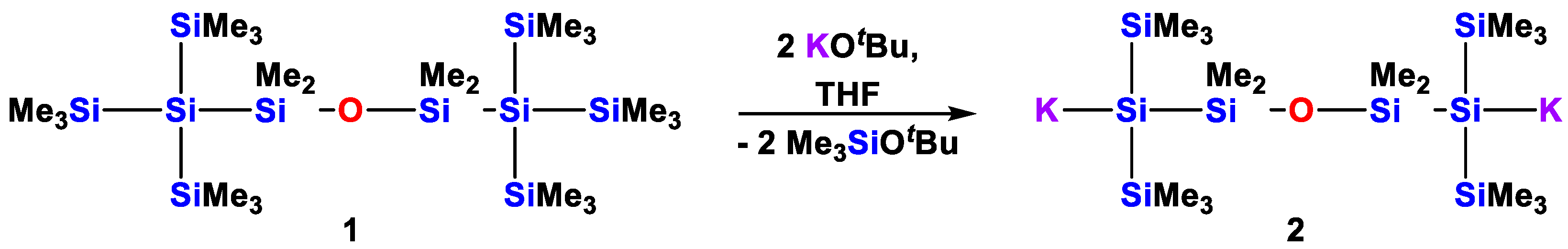{kind=link}
{kind=link}
{kind=link}
{kind=link}
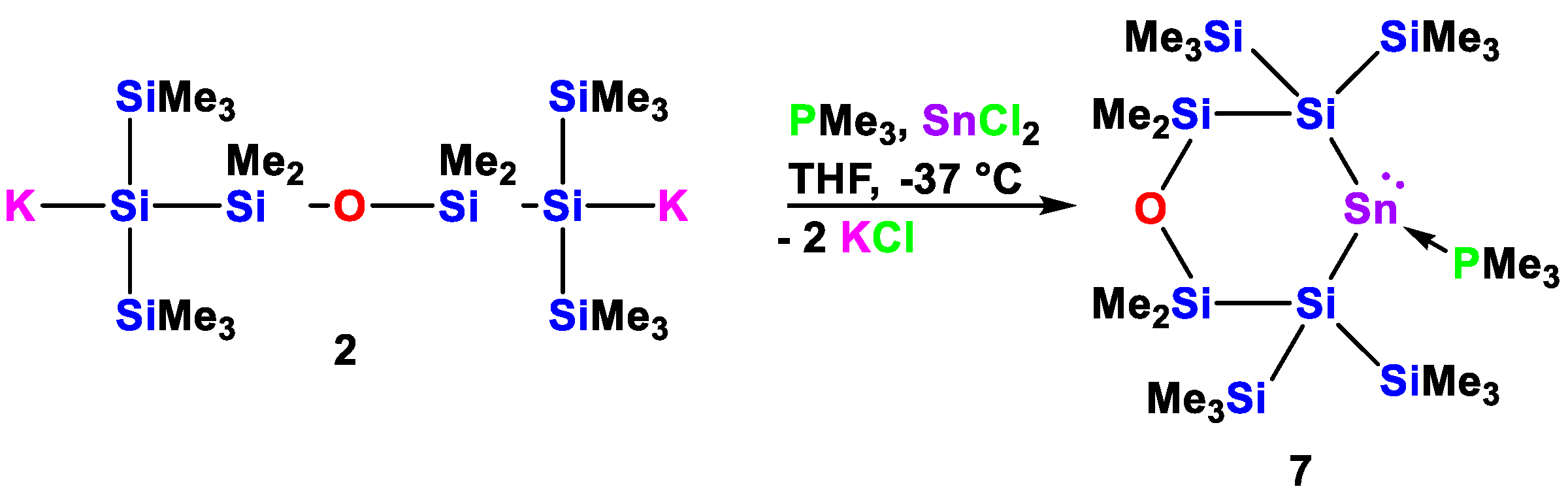{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
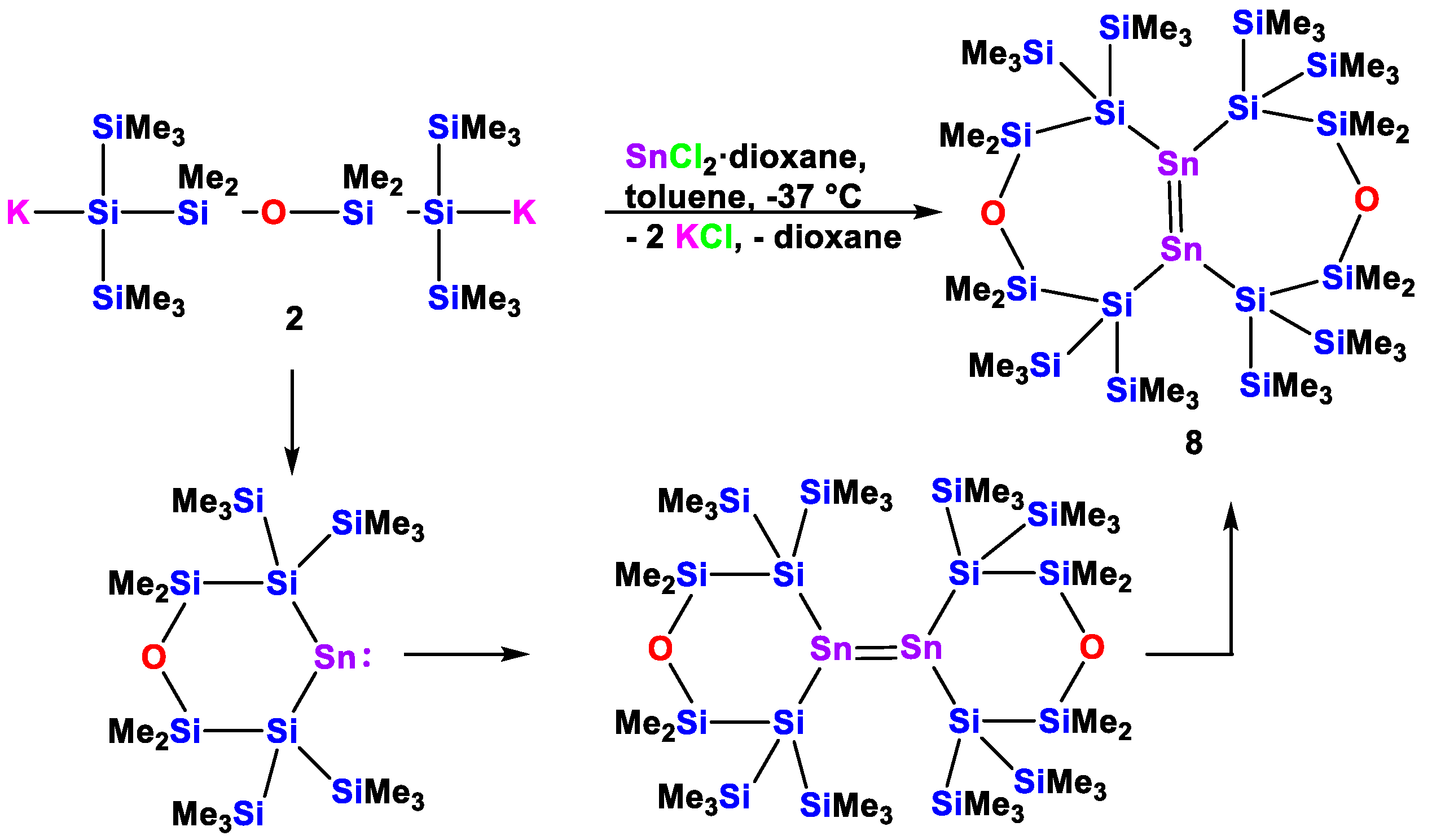
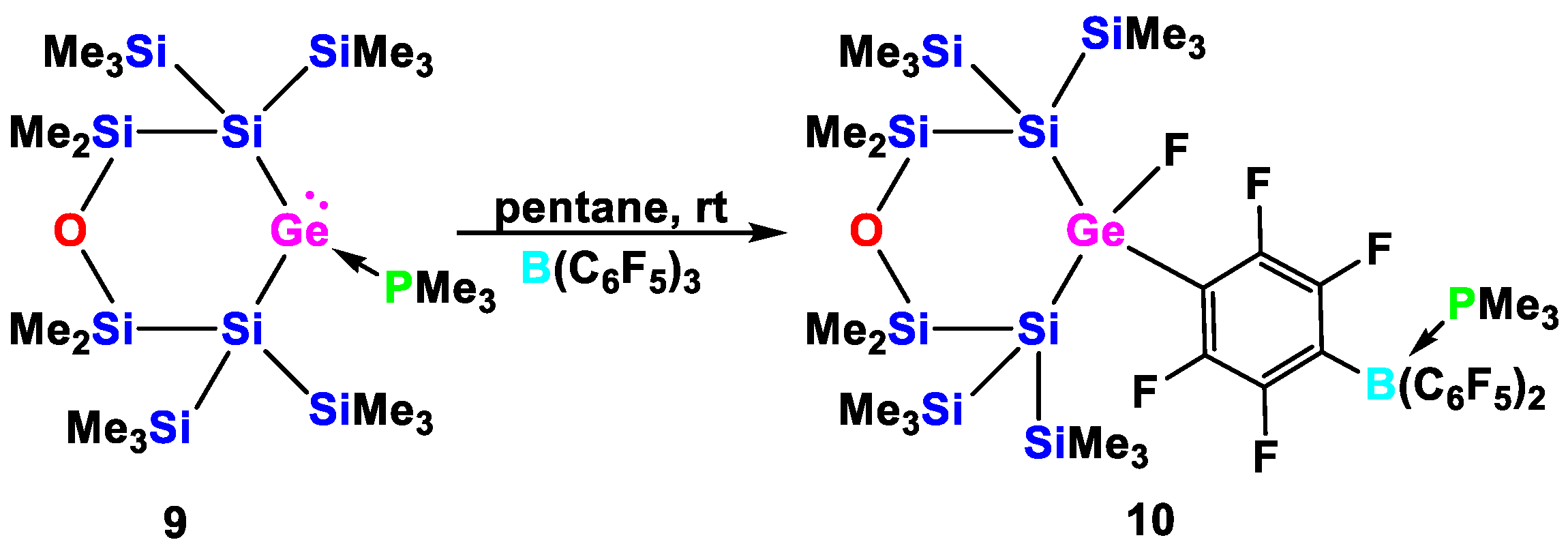
2. Results and Discussion
3. Experimental Section
3.1. General Remarks
3.2. X-ray Structure Determination
3.3. Synthesis
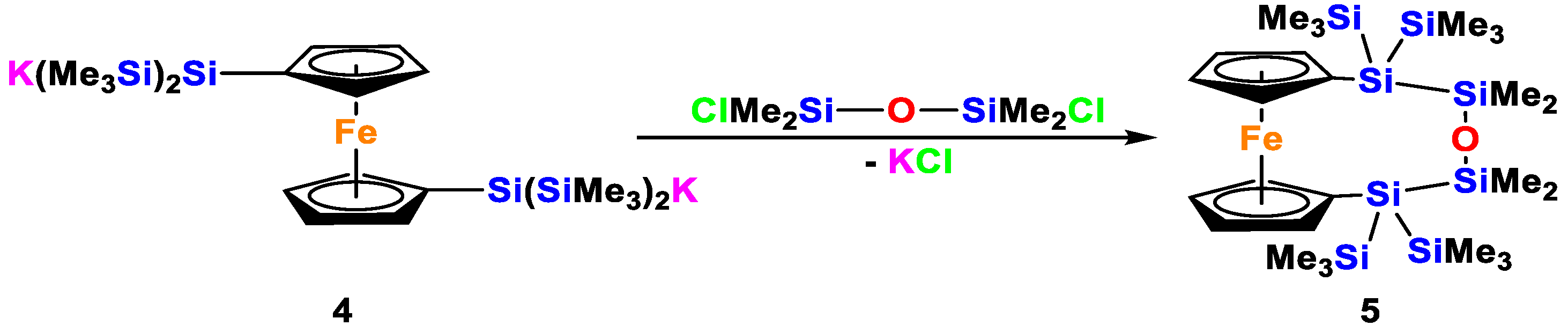
3.3.1. 1,1′-ansa-[2,2,4,4-Tetramethyl-1,1,5,5-tetrakis(trimethylsilyl)-3-oxa-1,2,4,5-tetrasila-1,5-diyl]ferrocene (5)
3.3.2. 1,1’-ansa-[1,5-Dipotassio-2,2,4,4-tetramethyl-1,5-bis(trimethylsilyl)-3-oxa-1,2,4,5-tetrasila-1,5-diyl]ferrocene (6)
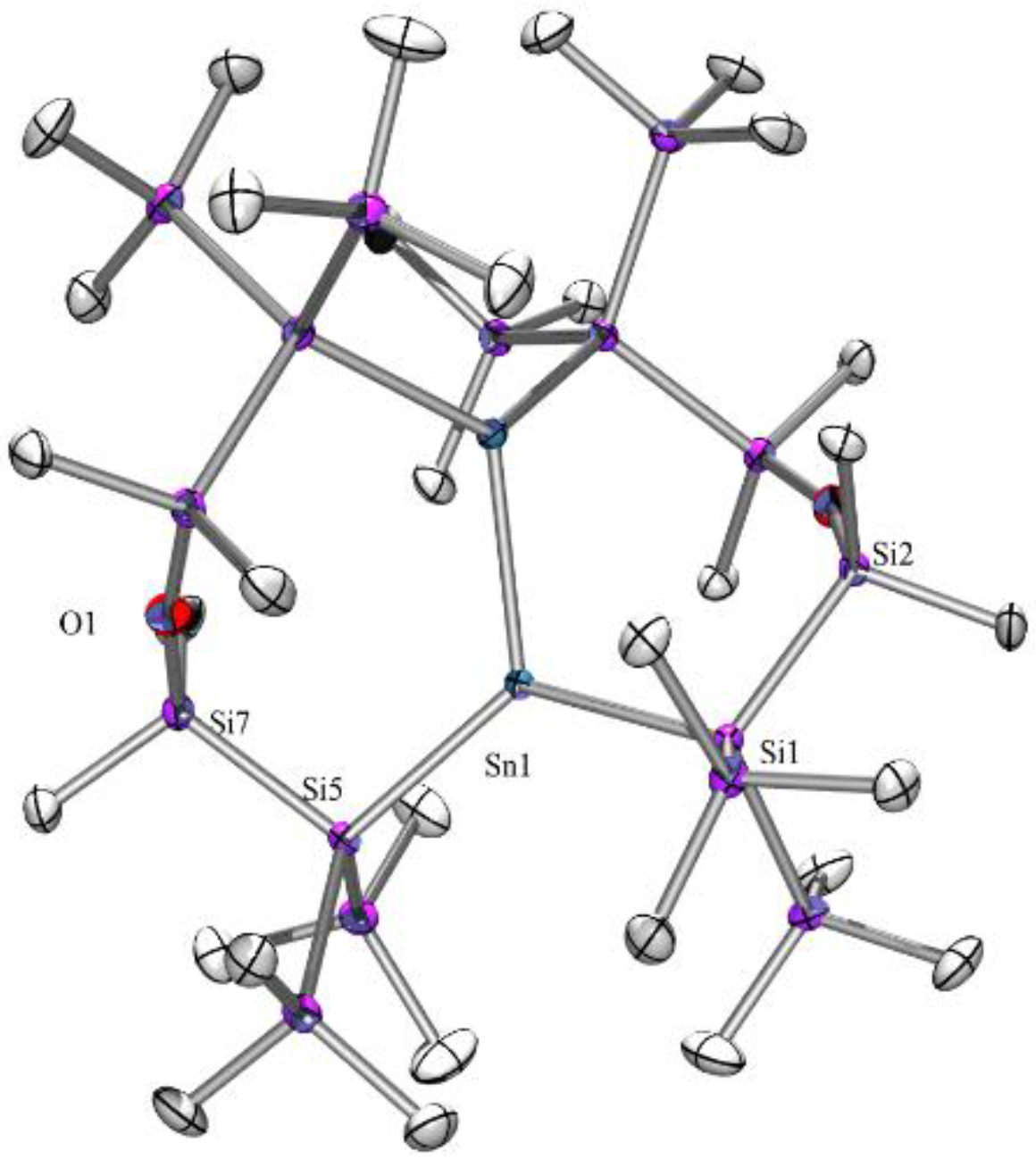
3.3.3. 2-Stanna-5-oxa-1,1,3,3-tetrakis(trimethylsilyl)tetramethylcyclohexasilan-2-ylidene·PMe3 (7)
3.3.4. 1,7-Distanna-2,2,6,6,8,8,12,12-octakis(trimethylsilyl)-4,10-dioxabicyclo[5.5.0]-dodecasil-1,7-ene (8)
3.3.5. 2-Germa-5-oxa-1,1,3,3-tetrakis(trimethylsilyl)tetramethylcyclohexasilan-2-ylidene·PMe3 (9)
3.3.6. C-F Insertion Product from the Reaction of 9 with B(C6F5)3 (10)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Liu, Y.; Onodera, K.; Takeda, N.; Ouali, A.; Unno, M. Synthesis and Characterization of Functionalizable Silsesquioxanes with Ladder-type Structures. Organometallics 2019, 38, 4373–4376. [Google Scholar] [CrossRef]
- Dankert, F.; Weigend, F.; von Hänisch, C. Not Non-Coordinating at All: Coordination Compounds of the Cyclodimethylsiloxanes Dn (D = Me2SiO; n = 6, 7) and Group 2 Metal Cations. Inorg. Chem. 2019, 58, 15417–15422. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Shiba, H.; Yoshikawa, M.; Wada, H.; Shimojima, A.; Kuroda, K. Synthesis of Polycyclic and Cage Siloxanes by Hydrolysis and Intramolecular Condensation of Alkoxysilylated Cyclosiloxanes. Chem. Eur. J. 2019, 25, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Oba, Y.; Nakajima, Y.; Shimada, S.; Sato, K. One-Pot Iterative Synthesis of Sequence-Controlled Oligosiloxanes. Angew. Chem. 2018, 130, 4727–4731. [Google Scholar] [CrossRef]
- Brook, M.A. New Control Over Silicone Synthesis Using SiH Chemistry: The Piers Rubinsztajn Reaction. Chem. Eur. J. 2018, 24, 8458–8469. [Google Scholar] [CrossRef]
- Yokouchi, Y.; Ishida, S.; Onodera, T.; Oikawa, H.; Iwamoto, T. Facile synthesis and bridgehead-functionalization of bicyclo[3.3.3]pentasiloxanes. Chem. Commun. 2017, 54, 268–270. [Google Scholar] [CrossRef]
- Thompson, D.B.; Brook, M.A. Rapid Assembly of Complex 3D Siloxane Architectures. J. Am. Chem. Soc. 2008, 130, 32–33. [Google Scholar] [CrossRef]
- Cui, C.; Qin, G. Controlled synthesis cyclosiloxanes by NHC-catalyzed hydrolytic oxidation of dihydrosilanes. Dalton Trans. 2017, 46, 8746–8750. [Google Scholar]
- Unno, M.; Tanaka, R.; Tanaka, S.; Takeuchi, T.; Kyushin, S.; Matsumoto, H. Oligocyclic Ladder Polysiloxanes: Alternative Synthesis by Oxidation. Organometallics 2005, 24, 765–768. [Google Scholar] [CrossRef]
- Marschner, C. Preparation and Reactions of Polysilanyl Anions and Dianions. Organometallics 2006, 25, 2110–2125. [Google Scholar] [CrossRef]
- Zitz, R.; Hlina, J.; Aghazadeh Meshgi, M.; Krenn, H.; Marschner, C.; Szilvási, T.; Baumgartner, J. Using Functionalized Silyl Ligands to Suppress Solvent Coordination to Silyl Lanthanide(II) Complexes. Inorg. Chem. 2017, 56, 5328–5341. [Google Scholar] [CrossRef] [PubMed]
- Zitz, R.; Hlina, J.; Arp, H.; Kinschel, D.; Marschner, C.; Baumgartner, J. Group 4 Metal and Lanthanide Complexes in the Oxidation State +3 with Tris(trimethylsilyl)silyl Ligands. Inorg. Chem. 2019, 58, 7107–7117. [Google Scholar] [CrossRef]
- Zitz, R.; Arp, H.; Hlina, J.; Walewska, M.; Marschner, C.; Szilvási, T.; Blom, B.; Baumgartner, J. Open-Shell Lanthanide(II+) or -(III+) Complexes Bearing σ-Silyl and Silylene Ligands: Synthesis, Structure, and Bonding Analysis. Inorg. Chem. 2015, 54, 3306–3315. [Google Scholar] [CrossRef] [PubMed]
- Zitz, R.; Hlina, J.; Gatterer, K.; Marschner, C.; Szilvási, T.; Baumgartner, J. Neutral “Cp-Free” Silyl-Lanthanide(II) Complexes: Synthesis, Structure, and Bonding Analysis. Inorg. Chem. 2015, 54, 7065–7072. [Google Scholar] [CrossRef] [PubMed]
- Zitz, R.; Baumgartner, J.; Marschner, C. Chemistry of a 1,5-Oligosilanylene Dianion Containing a Disiloxane Unit. Organometallics 2019, 38, 1159–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Juaid, S.S.; Derouiche, Y.; Hitchcock, P.B.; Lickiss, P.D. Preparation and X-ray crystal structure of an unusual bicyclic trisilane-tris(disoloxane). J. Organomet. Chem. 1988, 341, 241–245. [Google Scholar] [CrossRef]
- Jäger-Fiedler, U.; Köckerling, M.; Reinke, H.; Krempner, C. Discrete oxygen containing oligosilane dendrimers-modelling oxygen defects in silicon nanomaterials. Chem. Commun. 2010, 46, 4535–4537. [Google Scholar] [CrossRef]
- Krempner, C.; Jager-Fiedler, U.; Kockerling, M.; Reinke, H. Synthesis and Structures of Titanium and Zirconium Trisiloxides. Organometallics 2009, 28, 382–385. [Google Scholar] [CrossRef]
- Wagner, H.; Baumgartner, J.; Marschner, C. 1,1‘-Oligosilylferrocene Compounds. Organometallics 2007, 26, 1762–1770. [Google Scholar] [CrossRef]
- Fischer, R.; Konopa, T.; Ully, S.; Baumgartner, J.; Marschner, C. Route Si6 revisited. J. Organomet. Chem. 2003, 685, 79–92. [Google Scholar] [CrossRef]
- Mechtler, C.; Marschner, C. Polysilyldianions—Synthesis and reactivity. Tetrahedron Lett. 1999, 40, 7777–7778. [Google Scholar] [CrossRef]
- Kayser, C.; Kickelbick, G.; Marschner, C. Simple Synthesis of Oligosilyl-α,ω-dipotassium Compounds. Angew. Chem. Int. Ed. 2002, 41, 989–992. [Google Scholar] [CrossRef]
- Fischer, R.; Frank, D.; Gaderbauer, W.; Kayser, C.; Mechtler, C.; Baumgartner, J.; Marschner, C. α,ω-Oligosilyl Dianions and Their Application in the Synthesis of Homo- and Heterocyclosilanes. Organometallics 2003, 22, 3723–3731. [Google Scholar] [CrossRef]
- Fischer, R.; Konopa, T.; Baumgartner, J.; Marschner, C. Small Cyclosilanes: Syntheses and Reactions toward Mono- and Dianions. Organometallics 2004, 23, 1899–1907. [Google Scholar] [CrossRef]
- Mechtler, C.; Zirngast, M.; Gaderbauer, W.; Wallner, A.; Baumgartner, J.; Marschner, C. Synthesis of oligosilyldi- and trianions. J. Organomet. Chem. 2006, 691, 150–158. [Google Scholar] [CrossRef]
- Katir, N.; Matioszek, D.; Ladeira, S.; Escudié, J.; Castel, A. Stable N-Heterocyclic Carbene Complexes of Hypermetallyl Germanium(II) and Tin(II) Compounds. Angew. Chem. Int. Ed. 2011, 50, 5352–5355. [Google Scholar] [CrossRef]
- Klinkhammer, K. Dihypersilylstannylene and dihypersilylplumbylene—two Lewis-amphoteric carbene homologues. Polyhedron 2002, 21, 587–598. [Google Scholar] [CrossRef]
- Arp, H.; Marschner, C.; Baumgartner, J.; Zark, P.; Müller, T. Coordination Chemistry of Disilylated Stannylenes with Group 10 d10 Transition Metals: Silastannene vs Stannylene Complexation. J. Am. Chem. Soc. 2013, 135, 7949–7959. [Google Scholar] [CrossRef]
- Walewska, M.; Baumgartner, J.; Marschner, C. Synthesis of vinyl germylenes. Chem. Commun. 2015, 51, 276–278. [Google Scholar] [CrossRef]
- Arp, H.; Baumgartner, J.; Marschner, C.; Müller, T. A Cyclic Disilylated Stannylene: Synthesis, Dimerization, and Adduct Formation. J. Am. Chem. Soc. 2011, 133, 5632–5635. [Google Scholar] [CrossRef]
- Hlina, J.; Baumgartner, J.; Marschner, C.; Zark, P.; Müller, T. Coordination Chemistry of Disilylated Germylenes with Group 4 Metallocenes. Organometallics 2013, 32, 3300–3308. [Google Scholar] [CrossRef] [PubMed]
- Hlina, J.; Baumgartner, J.; Marschner, C.; Albers, L.; Müller, T.; Jouikov, V. Formation and Properties of a Bicyclic Silylated Digermene. Chem. Eur. J. 2014, 20, 9357–9366. [Google Scholar] [CrossRef] [PubMed]
- Walewska, M.; Hlina, J.; Gaderbauer, W.; Wagner, H.; Baumgartner, J.; Marschner, C. NHC Adducts of Disilylated Germylenes and Stannylenes and Their Coordination Chemistry with Group 11 Metals. Z. Anorg. Allg. Chem. 2016, 642, 1304–1313. [Google Scholar] [CrossRef]
- Klinkhammer, K.W.; Schwarz, W. Bis(hypersilyl)tin and Bis(hypersilyl)lead, Two Electron-Rich Carbene Homologs. Angew. Chem. Int. Ed. Engl. 1995, 34, 1334–1336. [Google Scholar] [CrossRef]
- Fukawa, T.; Lee, V.Ya.; Nakamoto, M.; Sekiguchi, A. Tetrakis(di-tert-butylmethylsilyl)distannene and Its Anion Radical. J. Am. Chem. Soc. 2004, 126, 11758–11759. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh Meshgi, M.; Zitz, R.; Walewska, M.; Baumgartner, J.; Marschner, C. Tuning the Si–N Interaction in Metalated Oligosilanylsilatranes. Organometallics 2017, 36, 1365–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hlina, J.; Baumgartner, J.; Marschner, C.; Albers, L.; Müller, T. Cyclic Disilylated and Digermylated Germylenes. Organometallics 2013, 32, 3404–3410. [Google Scholar] [CrossRef]
- Walewska, M.; Hlina, J.; Baumgartner, J.; Müller, T.; Marschner, C. Basic Reactivity Pattern of a Cyclic Disilylated Germylene. Organometallics 2016, 35, 2728–2737. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, P.P.; Xiao, M.; Pae, D.H.; Berger, D.J.; Haile, T.; Chen, T.; Lei, D.; Winchester, W.R.; Jiang, P. The quest for triplet ground state silylenes. J. Organomet. Chem. 2002, 646, 68–79. [Google Scholar] [CrossRef]
- Lawson, J.R.; Melen, R.L. Tris(pentafluorophenyl)borane and Beyond: Modern Advances in Borylation Chemistry. Inorg. Chem. 2017, 56, 8627–8643. [Google Scholar] [CrossRef] [Green Version]
- Swamy, V.S.V.S.N.; Parvin, N.; Raj, K.V.; Vanka, K.; Sen, S.S. C(sp3)–F, C(sp2)–F and C(sp3)–H bond activation at silicon(II) centers. Chem. Commun. 2017, 53, 9850–9853. [Google Scholar] [CrossRef] [PubMed]
- Jana, A.; Samuel, P.P.; Tavčar, G.; Roesky, H.W.; Schulzke, C. Selective Aromatic C-F and C-H Bond Activation with Silylenes of Different Coordinate Silicon. J. Am. Chem. Soc. 2010, 132, 10164–10170. [Google Scholar] [CrossRef] [PubMed]
- Samuel, P.P.; Singh, A.P.; Sarish, S.P.; Matussek, J.; Objartel, I.; Roesky, H.W.; Stalke, D. Oxidative Addition Versus Substitution Reactions of Group 14 Dialkylamino Metalylenes with Pentafluoropyridine. Inorg. Chem. 2013, 52, 1544–1549. [Google Scholar] [CrossRef] [PubMed]
- Mondal, T.; De, S.; Koley, D. DFT Study on C–F Bond Activation by Group 14 Dialkylamino Metalylenes: A Competition between Oxidative Additions versus Substitution Reactions. Inorg. Chem. 2017, 56, 10633–10643. [Google Scholar] [CrossRef] [PubMed]
- Chase, P.A.; Parvez, M.; Piers, W.E. Trimethylphosphine-tris(pentafluorophenyl)borane. Acta Cryst. E 2006, 62, o5181–o5183. [Google Scholar] [CrossRef]
- Ullrich, M.; Lough, A.J.; Stephan, D.W. Reversible, Metal-Free, Heterolytic Activation of H2 at Room Temperature. J. Am. Chem. Soc. 2009, 131, 52–53. [Google Scholar] [CrossRef]
- Hassler, K.; Dzambaski, A.; Baumgartner, J. Dihaloheptasilanes X2Si[SiMe(SiMe3)2]2 as potential precursors for silylenes, disilenes and cyclotrisilanes. Silicon Chem. 2007, 3, 271–288. [Google Scholar] [CrossRef]
- Pangborn, A.B.; Giardello, M.A.; Grubbs, R.H.; Rosen, R.K.; Timmers, F.J. Safe and Convenient Procedure for Solvent Purification. Organometallics 1996, 15, 1518–1520. [Google Scholar] [CrossRef]
- Morris, G.A.; Freeman, R. Enhancement of Nuclear Magnetic Resonance Signals by Polarization Transfer. J. Am. Chem. Soc. 1979, 101, 760–762. [Google Scholar] [CrossRef]
- Helmer, B.J.; West, R. Enhancement of 29Si NMR Signals by Proton Polarization Transfer. Organometallics 1982, 1, 877–879. [Google Scholar] [CrossRef]
- Bruker-AXS. SAINTPLUS: Software Reference Manual; Version 6.45; Bruker-AXS: Madison, WI, USA, 2003. [Google Scholar]
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Cryst. A 1995, 51, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SADABS; Version 2.10; Bruker AXS Inc.: Madison, WI, USA, 2003. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Available online: http://www.povray.org/download/ (accessed on 9 July 2008).
Sample Availability: Samples of the compounds are not available from the authors. |
















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zitz, R.; Pöcheim, A.; Baumgartner, J.; Marschner, C. A 1,5-Oligosilanylene Dianion as Building Block for Oligosiloxane Containing Cages, Ferrocenophanes, and Cyclic Germylenes and Stannylenes. Molecules 2020, 25, 1322. https://doi.org/10.3390/molecules25061322
Zitz R, Pöcheim A, Baumgartner J, Marschner C. A 1,5-Oligosilanylene Dianion as Building Block for Oligosiloxane Containing Cages, Ferrocenophanes, and Cyclic Germylenes and Stannylenes. Molecules. 2020; 25(6):1322. https://doi.org/10.3390/molecules25061322
Chicago/Turabian StyleZitz, Rainer, Alexander Pöcheim, Judith Baumgartner, and Christoph Marschner. 2020. "A 1,5-Oligosilanylene Dianion as Building Block for Oligosiloxane Containing Cages, Ferrocenophanes, and Cyclic Germylenes and Stannylenes" Molecules 25, no. 6: 1322. https://doi.org/10.3390/molecules25061322
APA StyleZitz, R., Pöcheim, A., Baumgartner, J., & Marschner, C. (2020). A 1,5-Oligosilanylene Dianion as Building Block for Oligosiloxane Containing Cages, Ferrocenophanes, and Cyclic Germylenes and Stannylenes. Molecules, 25(6), 1322. https://doi.org/10.3390/molecules25061322