
Diaryltin Dihydrides and Aryltin Trihydrides with Intriguing Stability



Abstract
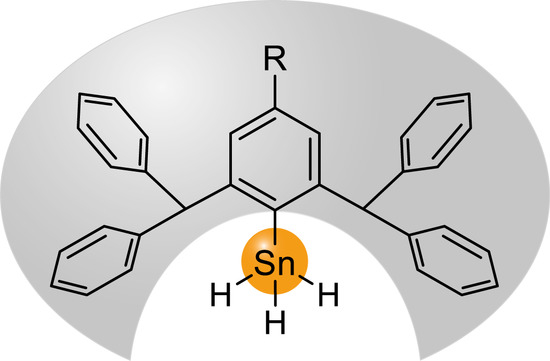
:
1. Introduction
2. Results
2.1. Synthesis and Spectroscopic Data
2.2. Solid State Structures
3. Materials and Methods
3.1. Synthesis
3.1.1. iPrAr*I (1)
3.1.2. MeAr*I (2)
3.1.3. General Procedure for Ar*Li
iPrAr*Li (3)
MeAr*Li (4)
3.1.4. General Procedure for Ar*2SnCl2
iPrAr*2SnCl2 (5)
MeAr*2SnCl2 (6)
3.1.5. iPrAr*SnMe3 (7)
3.1.6. iPrAr*SnCl2Me (8)
3.1.7. General Procedure for Ar*SnI3
iPrAr*SnI3 (9)
MeAr*SnI3 (10)
3.1.8. General Procedure for Ar*2SnH2
iPrAr*2SnH2 (11)
MeAr*2SnH2 (12)
3.1.9. General Procedure for Ar*SnH3
iPrAr*SnH3 (13)
MeAr*SnH3 (14)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rivard, E. Group 14 Inorganic Hydrocarbon Analogues. Chem. Soc. Rev. 2016, 45, 989–1003. [Google Scholar] [CrossRef]
- Holleman, A.F.; Nils Wiberg, E.; Fischer, G. Lehrbuch Der Anorganischen Chemie; Walter de Gruyter: Berlin, Germanny, 2007. [Google Scholar] [CrossRef]
- Aldridge, S.; Downs, A.J. Hydrides of the Main-Group Metals: New Variations on an Old Theme. Chem. Rev. 2001, 101, 3305–3365. [Google Scholar] [CrossRef]
- Davies, A.G. Organotin Hydrides. In Organotin Chemistry; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2004; pp. 245–265. [Google Scholar]
- Elschenbroich, C. Organometallchemie; Badura, B., Schröder, H., Vetter, C., Eds.; Vieweg+Teubner: Wiesbaden, Germany, 2008. [Google Scholar] [CrossRef]
- Braunstein, P.; Morise, X. Dehydrogenative Coupling of Hydrostannanes Catalyzed by Transition Metal Complexes. Chem. Rev. 2000, 100, 3542–3552. [Google Scholar] [CrossRef]
- Hill, M.S. Homocatenation of Metal and Metalloid Main Group Elements. In Metal-Metal Bonding; Parking, G., Ed.; Springer-Verlag: Berlin, Germany, 2010; Volume 136, pp. 189–216. [Google Scholar] [CrossRef]
- Caseri, W. Polystannanes: Processible Molecular Metals With. Chem. Soc. Rev. 2016, 45, 5187–5199. [Google Scholar] [CrossRef]
- Tokitoh, N.; Suzuki, H.; Matsumoto, T.; Matsuhashi, Y.; Okazaki, R.; Goto, M. 1,2,3,4,5-Tetrathiametallolanes of Group 14 Metals, RR′MS4 (M = Si, Ge, and Sn): Synthesis and Crystal Structures. J. Am. Chem. Soc. 1991, 113, 7047–7049. [Google Scholar] [CrossRef]
- Saito, M.; Hashimoto, H.; Tajima, T.; Ikeda, M. Synthesis and Structures of Polychalcogenadistannabicyclo[k.l.m]Alkanes. J. Organomet. Chem. 2007, 692, 2729–2735. [Google Scholar] [CrossRef]
- Hayes, P.G.; Gribble, C.W.; Waterman, R.; Tilley, T.D. A Hydrogen-Substituted Osmium Stannylene Complex: Isomerization to a Metallostannylene Complex via an Unusual α-Hydrogen Migration from Tin to Osmium. J. Am. Chem. Soc. 2009, 131, 4606–4607. [Google Scholar] [CrossRef]
- Liu, H.J.; Guihaumé, J.; Davin, T.; Raynaud, C.; Eisenstein, O.; Tilley, T.D. 1,2-Hydrogen Migration To a Saturated Ruthenium Complex Via Reversal of Electronic Properties for Tin in a Stannylene-To-Metallostannylene Conversion. J. Am. Chem. Soc. 2014, 136, 13991–13994. [Google Scholar] [CrossRef]
- Sindlinger, C.P.; Wesemann, L. Hydrogen Abstraction from Organotin Di- and Trihydrides by N-Heterocyclic Carbenes: A New Method for the Preparation of NHC Adducts to Tin(I) Species and Observation of an Isomer of a Hexastannabenzene Derivative [R6Sn6]. Chem. Sci. 2014, 5, 2739–2746. [Google Scholar] [CrossRef]
- Sindlinger, C.P.; Weiß, S.; Schubert, H.; Wesemann, L. Nickel-Triad Complexes of a Side-on Coordinating Distannene. Angew. Chem.-Int. Ed. 2015, 54, 4087–4091. [Google Scholar] [CrossRef] [PubMed]
- Maudrich, J.J.; Sindlinger, C.P.; Aicher, F.S.W.; Eichele, K.; Schubert, H.; Wesemann, L. Reductive Elimination of Hydrogen from Bis(Trimethylsilyl)Methyltin Trihydride and Mesityltin Trihydride. Chem.-A Eur. J. 2017, 23, 2192–2200. [Google Scholar] [CrossRef] [PubMed]
- Diab, F.; Aicher, F.S.W.; Sindlinger, C.P.; Eichele, K.; Schubert, H.; Wesemann, L. Reductive Elimination and Oxidative Addition of Hydrogen at Organostannylium and Organogermylium Cations. Chem.-A Eur. J. 2019, 25, 4426–4434. [Google Scholar] [CrossRef] [PubMed]
- Maudrich, J.J.; Diab, F.; Weiß, S.; Widemann, M.; Dema, T.; Schubert, H.; Krebs, K.M.; Eichele, K.; Wesemann, L. Deprotonation of Organogermanium and Organotin Trihydrides. Inorg. Chem. 2019, 58, 15758–15768. [Google Scholar] [CrossRef]
- Maudrich, J.J.; Widemann, M.; Diab, F.; Kern, R.H.; Sirsch, P.; Sindlinger, C.P.; Schubert, H.; Wesemann, L. Hydridoorganostannylene Coordination: Group 4 Metallocene Dichloride Reduction in Reaction with Organodihydridostannate Anions. Chem.-A Eur. J. 2019, 25, 16081–16087. [Google Scholar] [CrossRef] [Green Version]
- Kraus, C.A.; Greer, W.N. The Preparation and Properties of Trimethylstannane. J. Am. Chem. Soc. 1922, 44, 2629–2633. [Google Scholar] [CrossRef] [Green Version]
- Finholt, A.E.; Bond, A.C.; Wilzbach, K.E.; Schlesinger, H.I. The Preparation and Some Properties of Hydrides of Elements of the Fourth Group of the Periodic System and of Their Organic Derivatives. J. Am. Chem. Soc. 1947, 69, 2692–2696. [Google Scholar] [CrossRef]
- Neumann, W.P.; Niermann, H. Darstellung Von Organozinn-Mono-, -Di- Und -Tri-Hydriden. Eur. J. Inorg. Chem. 1962, 653, 164–172. [Google Scholar]
- Bresien, J.; Goicoechea, J.M.; Hinz, A.; Scharnhölz, M.T.; Schulz, A.; Suhrbier, T.; Villinger, A. Increasing Steric Demand through Flexible Bulk-Primary Phosphanes with 2,6-Bis(Benzhydryl)Phenyl Backbones. Dalton Trans. 2019, 48, 3786–3794. [Google Scholar] [CrossRef]
- Protchenko, A.V.; Birjkumar, K.H.; Dange, D.; Schwarz, A.D.; Vidovic, D.; Jones, C.; Kaltsoyannis, N.; Mountford, P.; Aldridge, S. A Stable Two-Coordinate Acyclic Silylene. J. Am. Chem. Soc. 2012, 134, 6500–6503. [Google Scholar] [CrossRef]
- Hadlington, T.J.; Hermann, M.; Li, J.; Frenking, G.; Jones, C. Activation of H2 by a Multiply Bonded Amido-Digermyne: Evidence for the Formation of a Hydrido-Germylene. Angew. Chem. Int. Ed. 2013, 52, 10199–10203. [Google Scholar] [CrossRef] [PubMed]
- Hadlington, T.J.; Hermann, M.; Frenking, G.; Jones, C. Low Coordinate Germanium(II) and Tin(II) Hydride Complexes: Efficient Catalysts for the Hydroboration of Carbonyl Compounds. J. Am. Chem. Soc. 2014, 136, 3028–3031. [Google Scholar] [CrossRef] [PubMed]
- De Bruin-Dickason, C.N.; Boutland, A.J.; Dange, D.; Deacon, G.B.; Jones, C. Redox Transmetallation Approaches to the Synthesis of Extremely Bulky Amido-Lanthanoid(II) and -Calcium(II) Complexes. Dalton Trans. 2018, 47, 9512–9520. [Google Scholar] [CrossRef] [PubMed]
- Vrána, J.; Samsonov, M.A.; Němec, V.; Růžička, A. Access to the Most Sterically Crowded Anilines via Non-Catalysed C–C Coupling Reactions. Chem. Commun. 2020, 56, 2–5. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.; Mahon, M.F.; Molloy, K.C. Sterically Hindered Organotin Compounds II. Synthesis of the Organotin Halides (Mesityl)2SnXnY2-n, (Mesityl)SnXnY3-n and R4Sn2X2 (R = 2,4,6-IPr3C6H2, X = Cl, Y = Br). Crystal Structure of the Ditin Species (R2BrSn)2. J. Organomet. Chem. 1992, 435, 265–273. [Google Scholar] [CrossRef]
- Elhamzaoui, H.; Jousseaume, B.; Toupance, T.; Allouchi, H. α,ω-Bis(Trialkynyltin) Compounds with a Linear or Cross-Shaped Spacer. Organometallics 2007, 26, 3908–3917. [Google Scholar] [CrossRef]
- Prabusankar, G.; Jousseaume, B.; Toupance, T.; Allouchi, H. A Discrete Unsymmetrically Substituted Dihydrodioxadistannetane with Both Η1 and Intramolecular Η2μ2 Sulfonate Bondings. J. Organomet. Chem. 2008, 693, 3383–3386. [Google Scholar] [CrossRef]
- Bouska, M.; Dostá, L.; Jirásko, R.; Růžička, A.; Jambor, R. Synthesis of [{2,6-(Me2NCH2)2C 6H3}Sn(OH)0]6: An N-Sn Coordinated Stannonic Acid. Organometallics 2009, 28, 4258–4261. [Google Scholar] [CrossRef]
- Steller, B.G.; Fischer, R.C. Selective Preparation of Sterically Encumbered Diaryltin Dihalides from Grignard Reagents via Salt Metathesis and Halide Exchange. Eur. J. Inorg. Chem. 2019, 2019, 2591–2597. [Google Scholar] [CrossRef]
- Zeppek, C.; Pichler, J.; Torvisco, A.; Flock, M.; Uhlig, F. Aryltin Chlorides and Hydrides: Preparation, Detailed NMR Studies and DFT Calculations. J. Organomet. Chem. 2013, 740, 41–49. [Google Scholar] [CrossRef]
- Kapoor, R.N.; Apodaca, P.; Montes, M.; Gomez, F.D.; Pannell, K.H. Mixed Aryl-Alkyl Organotin Compounds, ArMeSnCl3-n (Ar = RC6H4, R = H, Ethyl, i-Propyl, t-Butyl; n-Hexyl, n-Octyl) and the Effect of R upon Antibiotic Activity. Appl. Organomet. Chem. 2005, 19, 518–522. [Google Scholar] [CrossRef]
- Pejchal, V.; J, H.; Lyčka, A. 13C and 119Sn NMR Spectra of Some Monophenyltin(IV) Compounds. Sci. Pap. Univ. Pardubice 1996, 2, 35–46. [Google Scholar]
- Novák, P.; Padělková, Z.; Císařová, I.; Kolářová, L.; Růžička, A.; Holeček, J. Structural Study of C,N-Chelated Monoorganotin(IV) Halides. Appl. Organomet. Chem. 2006, 20, 226–232. [Google Scholar] [CrossRef]
- Greene, B.P.T.; Bryan, R.F. Crystal Structure of Dichloro(Diphenyl)Tin. J. Chem. Soc. 1971, 2549–2554. [Google Scholar] [CrossRef]
- Kräuter, T.; Neumüller, B. Die Kristallstrukturen von MesSn(Cl)Me2 Und Mes2SnCl2. Zeitschrift für Naturforsch. B 1998, 53, 503–506. [Google Scholar] [CrossRef]
- Sharma, H.K.; Cervantes-lee, F.; Mahmoud, J.S.; Pannell, K.H. (Bis{2,4,6-Triisopropylphenyl}stannylene)Feroccenophane and Related Ring-Opened Products. Organometallics 1999, 18, 399–403. [Google Scholar] [CrossRef]
- Weidenbruch, M.; Schäfers, K.; Pohl, S.; Saak, W.; Peters, K.; von Schnering, H.G. Verbindungen Des Germaniums Und Zinns II *. Bildung Und Strukturen Isomerer “Dichlordisupermesitylstannane.”. J. Organomet. Chem. 1988, 346, 171–180. [Google Scholar] [CrossRef]
- Available online: https://www.ccdc.cam.ac.uk (accessed on 14 February 2020).
- Ahmad, S.U.; Beckmann, J.; Duthie, A. New Insights into the Formation and Reactivity of Molecular Organostannonic Acids. Chem. Asian J. 2010, 5, 160–168. [Google Scholar] [CrossRef]
- Johnson, B.P.; Almstätter, S.; Dielmann, F.; Bodensteiner, M.; Scheer, M. Synthesis and Reactivity of Low-Valent Group 14 Element Compounds. Z. Anorg. Allg. Chem. 2010, 636, 1275–1285. [Google Scholar] [CrossRef] [Green Version]
- Zeppek, C. Amine Base Induced Polymerization of Aryltin Hydrides: Mechanistic Insights & Nanomaterial Characterization. Ph.D. Thesis, Graz University of Technology, Graz, 2015. [Google Scholar]
- Schittelkopf, K.; Fischer, R.C.; Meyer, S.; Wilfling, P.; Uhlig, F. Catalytic Dehydrogenative Coupling of Diorganotindihydrides by Lanthanide Diamide Complexes†. Appl. Organomet. Chem. 2010, 24, 897–901. [Google Scholar] [CrossRef]
- Sindlinger, C.P.; Stasch, A.; Bettinger, H.F.; Wesemann, L. A Nitrogen-Base Catalyzed Generation of Organotin(Ii) Hydride from an Organotin Trihydride under Reductive Dihydrogen Elimination. Chem. Sci. 2015, 6, 4737–4751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthon-Gelloz, G.; Siegler, M.A.; Spek, A.L.; Tinant, B.; Reek, J.N.H.; Markó, I.E. IPr* an Easily Accessible Highly Hindered N-Heterocyclic Carbene. Dalton Trans. 2010, 39, 1444–1446. [Google Scholar] [CrossRef] [PubMed]
- Blessing, R.H. An Empirical Correction for Absorption Anisotropy. Acta Crystallogr. Sect. A 1995, A51, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SADABS Version 2.10 Siemens Area Detector Correction.; Universitaet Goettingen: Goettingen, Germany, 2003. [Google Scholar]
- Sheldrick, G.M. SHELXTL Version 6.1. Bruker AXS, Inc.: Wisconsin, WI, USA,, 2002.
- Sheldrick, G.M. GM SHELXS97 and SHELXL97; Universitaet Goettingen: Goettingen, Germany, 2002. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. Single-Crystal Structure Validation with the Program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 119Sn NMR (ppm) | 119Sn NMR (ppm) | 119Sn NMR (ppm) | 1J (1H, 117/119Sn) (Hz) | |
|---|---|---|---|---|
| X = Cl | X = I | X = H | ||
| iPrAr*2SnX2 | −65.96 | −498.22 * | −331.30 | 1942/2033 |
| MeAr*2SnX2 | −64.65 | −496.98 * | −331.51 | 1930/2019 |
| iPrAr*SnMe3 | −56.80 | - | - | - |
| iPrAr*SnMeX2 | +15.07 | - | - | - |
| iPrAr*SnX3 | - | −937.27 | −407.06 | 1843/1930 |
| MeAr*SnX3 | - | −939.57 | −406.67 | 1845/1931 |
| Sn-C (Å) | Sn-X (Å) | C-Sn-C (°) | X-Sn-X (°) | |
|---|---|---|---|---|
| iPrAr*2SnCl2 (5) | 2.1501(15) | 2.3781(5) | 125.77(7) | 94.49(3) |
| MeAr*2SnCl2 (6) | 2.155(5), 2.159(6) | 2.402(2), 2.344(2) | 119.5(2) | 95.82(5) |
| iPrAr*SnMe3 (7) | 2.189(1) | - | 113.04(6) 111.75(6) 114.81(6) | - |
| iPrAr*SnMeCl2 (8) | 2.141(7) 2.140(6) | 2.373(2), 2.375(2), 2.369(2) 2.381(2) | 131.2(3) 130.0(3) | 92.47(7) |
| iPrAr*SnI3 (9) | 2.161(2) | 2.7130(4), 2.6964(4), 2.6721(4) | - | 97.31(1), 106.04(1), 107.66(1) |
| MeAr*SnI3 (10) | 2.158(3) | 2.6994(4), 2.6752(4) | - | 105.743(11), 95.665(12) |
| Sn-C (Å) | Sn-H (Å) | C-Sn-C (°) | H-Sn-H (°) | |
|---|---|---|---|---|
| iPrAr*2SnH2 (11) | 2.187(3), 2.171(3) | 1.71(3), 1.70(4) | 105.9(1) | 109(2) |
| MeAr*2SnH2 (12) | 2.188(2), 2.186(2) | 1.79(2), 1.80(3) | 109.49(8) | 100.7(9) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steller, B.G.; Doler, B.; Fischer, R.C. Diaryltin Dihydrides and Aryltin Trihydrides with Intriguing Stability. Molecules 2020, 25, 1076. https://doi.org/10.3390/molecules25051076
Steller BG, Doler B, Fischer RC. Diaryltin Dihydrides and Aryltin Trihydrides with Intriguing Stability. Molecules. 2020; 25(5):1076. https://doi.org/10.3390/molecules25051076
Chicago/Turabian StyleSteller, Beate G., Berenike Doler, and Roland C. Fischer. 2020. "Diaryltin Dihydrides and Aryltin Trihydrides with Intriguing Stability" Molecules 25, no. 5: 1076. https://doi.org/10.3390/molecules25051076
APA StyleSteller, B. G., Doler, B., & Fischer, R. C. (2020). Diaryltin Dihydrides and Aryltin Trihydrides with Intriguing Stability. Molecules, 25(5), 1076. https://doi.org/10.3390/molecules25051076