2-(Arylamino)-6-(trifluoromethyl)nicotinic Acid Derivatives: New HIV-1 RT Dual Inhibitors Active on Viral Replication

 ,
,  ,
, 


Abstract
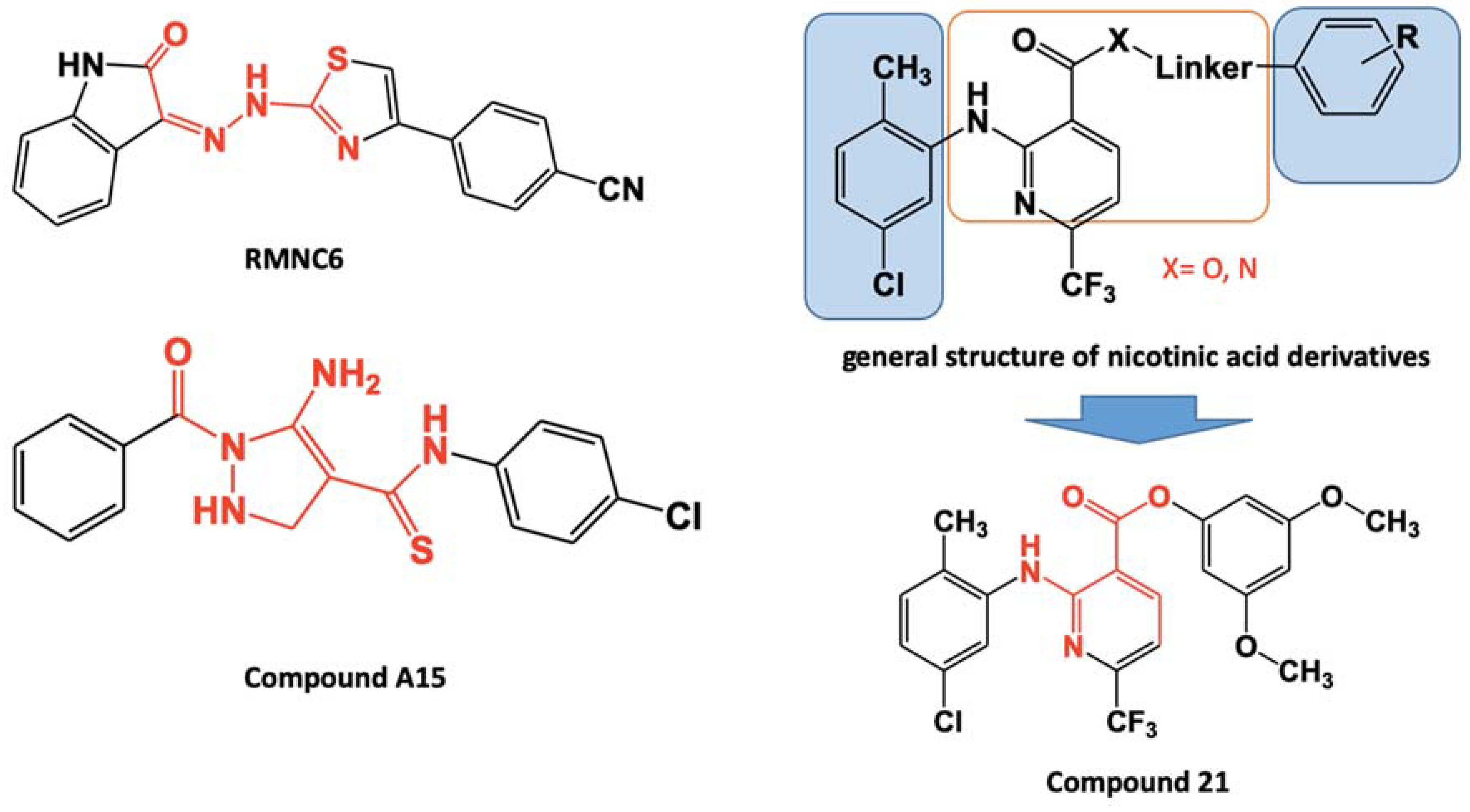
:1. Introduction
2. Results
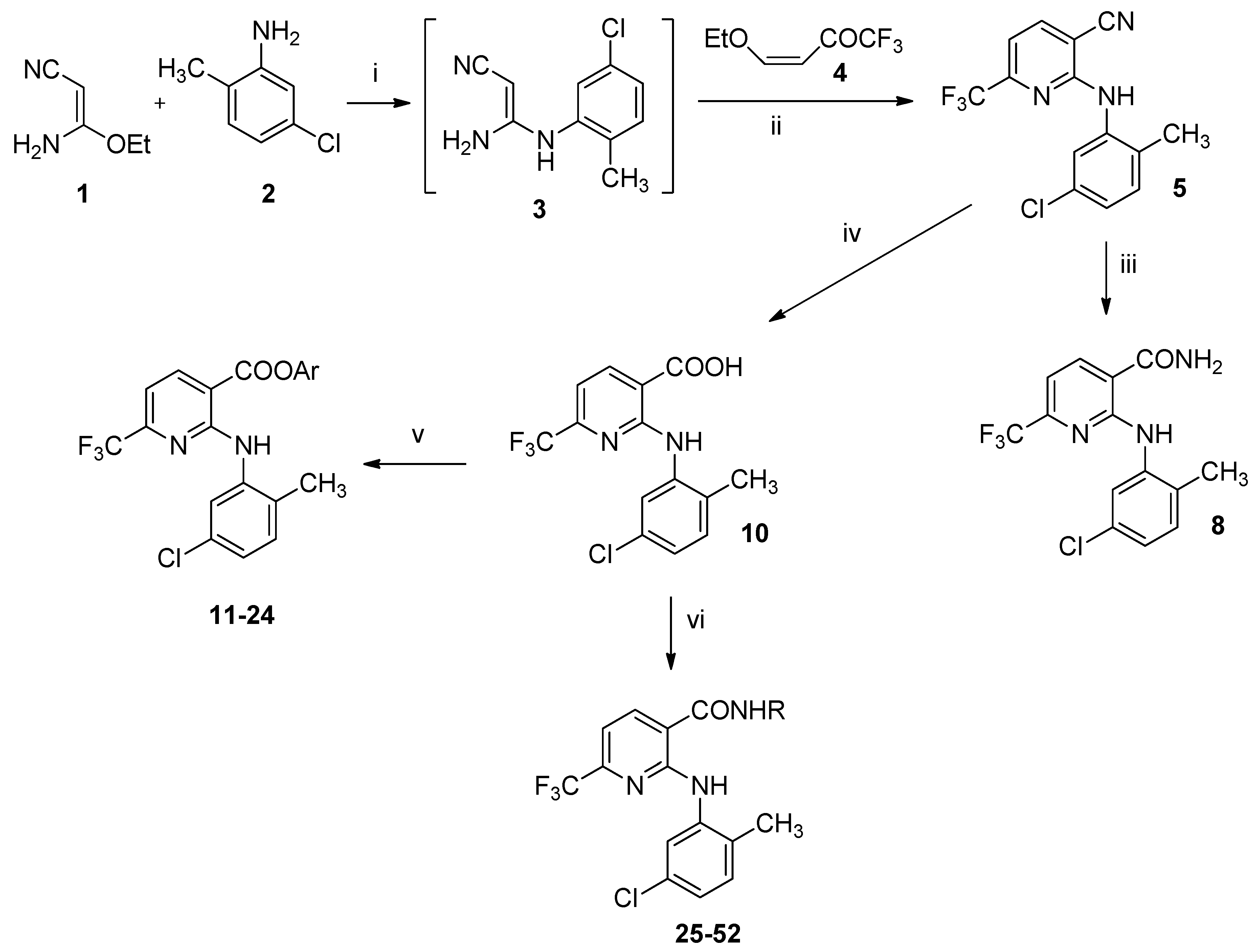
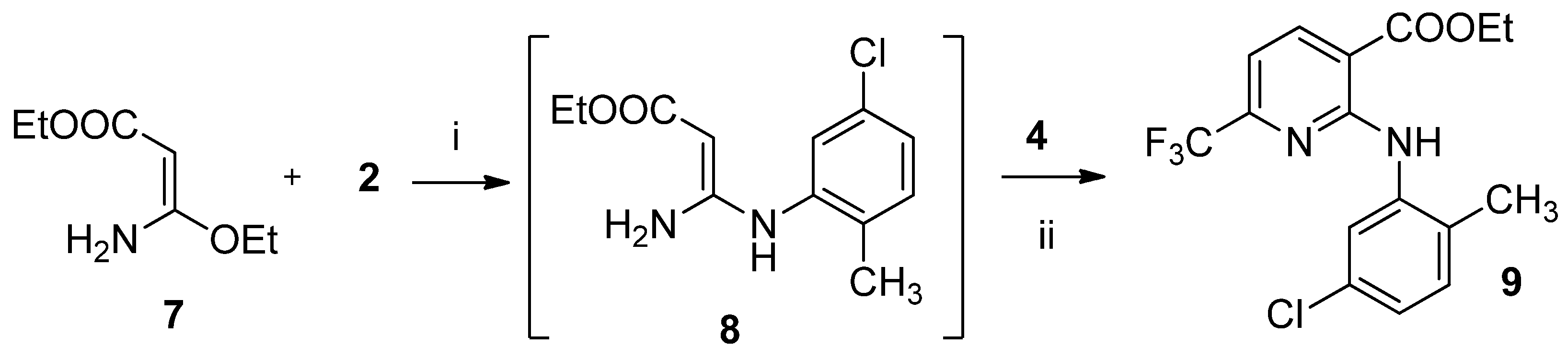
2.1. Synthesis of 2-amino-6-(trifluoromethyl)nicotinate derivatives
2.2. Evaluation of RNase H Inhibitory Activity
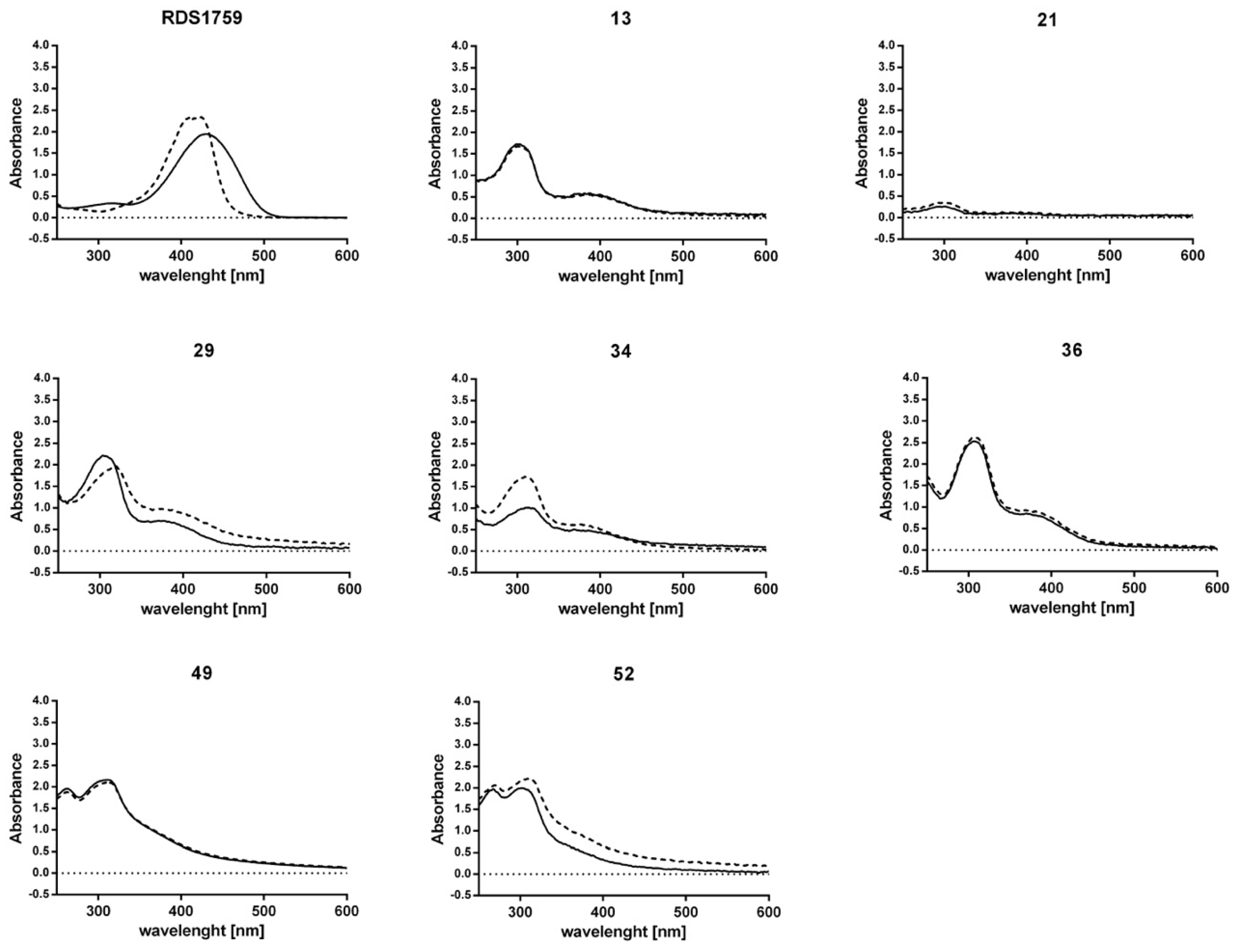
2.3. Evaluation of Antiviral Activity
2.4. Mode of Action Studies
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Procedure for the Synthesis of Esters (11–24)
4.1.2. General Procedure for the Synthesis of Amides (25–52)
4.2. Biology
4.2.1. Expression and Purification of Recombinant HIV-1 RTs Group M Subtype B
4.2.2. Mg2+ Coordination
4.2.3. Site-Directed Mutagenesis
4.2.4. HIV-1 DNA Polymerase-Independent RNase H Activity Determination
4.2.5. HIV-1 RNA-Dependent DNA Polymerase Activity Determination
4.2.6. Antiviral Assay
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- UNAIDS 2018 Global HIV Statistics. Available online: https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf (accessed on 17 February 2020).
- Fauci, A.S.; Folkers, G.K. Toward an AIDS-free generation. JAMA J. Am. Med. Assoc. 2012, 308, 343–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeks, S.G.; Lewin, S.R.; Havlir, D.V. The end of AIDS: HIV infection as a chronic disease. Lancet 2013, 382, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- Stella-Ascariz, N.; Arribas, J.R.; Paredes, R.; Li, J.Z. The role of HIV-1 drug-resistant minority variants in treatment failure. J. Infect. Dis. 2017, 216, S847–S850. [Google Scholar] [CrossRef] [PubMed]
- Gregson, J.; Tang, M.; Ndembi, N.; Hamers, R.L.; Marconi, V.C.; Brooks, K.; Theys, K.; Arruda, M.; Garcia, F.; Monge, S.; et al. Global epidemiology of drug resistance after failure of WHO recommended first-line regimens for adult HIV-1 infection: A multicentre retrospective cohort study. Lancet Infect. Dis. 2016, 16, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.; Corona, A.; Spöring, I.; Jordan, M.; Buchholz, B.; Maccioni, E.; Di Santo, R.; Bodem, J.; Tramontano, E.; Wöhrl, B.M. Biochemical characterization of a multi-drug resistant HIV-1 subtype AG reverse transcriptase: Antagonism of AZT discrimination and excision pathways and sensitivity to RNase H inhibitors. Nucleic Acids Res. 2016, 44, 2310–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Neogi, U. Following the path: Increasing trends of HIV-1 drug resistance in China. EClinicalMedicine 2020, 18, 100251. [Google Scholar] [CrossRef] [Green Version]
- Menéndez-Arias, L.; Berkhout, B. Retroviral reverse transcription. Virus Res. 2008, 134, 1–3. [Google Scholar] [CrossRef]
- Liu, S.; Abbondanzieri, E.A.; Rausch, J.W.; Le Grice, S.F.J.; Zhuang, X. Slide into action: Dynamic shuttling of HIV reverse transcriptase on nucleic acid substrates. Science 2008, 322, 1092–1097. [Google Scholar] [CrossRef]
- Krupovic, M.; Blomberg, J.; Coffin, J.M.; Dasgupta, I.; Fan, H.; Geering, A.D.; Gifford, R.; Harrach, B.; Hull, R.; Johnson, W.; et al. Ortervirales: New virus order unifying five families of reverse-transcribing viruses. J. Virol. 2018, 92, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.F.; Hostomska, Z.; Hostomsky, Z.; Jordan, S.R.; Matthews, D.A. Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase. Science 1991, 252, 88–95. [Google Scholar] [CrossRef]
- Tadokoro, T.; Kanaya, S. Ribonuclease H: Molecular diversities, substrate binding domains, and catalytic mechanism of the prokaryotic enzymes. FEBS J. 2009, 276, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Cherepanov, P. Retroviral integrase structure and DNA recombination mechanism. Microbiol. Spectr. 2014, 2, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyjek, M.; Figiel, M.; Nowotny, M. RNases H: Structure and mechanism. DNA Repair 2019, 102672. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.; Schneider, A.; Schweimer, K.; Rösch, P.; Wöhrl, B.M.; Tramontano, E. Inhibition of foamy virus reverse transcriptase by human immunodeficiency virus type 1 ribonuclease H inhibitors. Antimicrob. Agents Chemother. 2014, 58, 4086–4093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatz, O.; Cromme, F.V.; Naas, T.; Lindemann, D. Inactivation of the RNase H domain of HIV-1 reverse transcriptase blocks viral infectivity. Gene Regul. AIDS. 1990, 293–404. [Google Scholar]
- Tramontano, E.; Corona, A.; Menéndez-Arias, L. Ribonuclease H, an unexploited target for antiviral intervention against HIV and hepatitis B virus. Antivir. Res. 2019, 171, 104613. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.; Masaoka, T.; Tocco, G.; Tramontano, E.; Le Grice, S.F. Active site and allosteric inhibitors of the ribonuclease H activity of HIV reverse transcriptase. Futur. Med. Chem. 2013, 5, 2127–2139. [Google Scholar] [CrossRef]
- Wang, X.; Gao, P.; Menendez-Arias, L.; Liu, X.; Zhan, P.; Menéndez-Arias, L.; Liu, X.; Zhan, P. Update on recent developments in small molecular HIV-1 RNase H inhibitors (2013-2016): Opportunities and Challenges. Curr. Med. Chem. 2018, 25, 1682–1702. [Google Scholar] [CrossRef]
- Corona, A.; Esposito, F.; Tramontano, E. Can the ever-promising target HIV reverse transcriptase-associated RNase H become a success story for drug development? Future Virol. 2014, 9, 445–448. [Google Scholar] [CrossRef]
- Xi, Z.; Wang, Z.; Sarafianos, S.G.; Myshakina, N.S.; Ishima, R. Determinants of active-site inhibitor interaction with HIV-1 RNase H. ACS Infect. Dis. 2019, 5, 1963–1974. [Google Scholar] [CrossRef]
- Wang, L.; Tang, J.; Huber, A.D.; Casey, M.C.; Kirby, K.A.; Wilson, D.J.; Kankanala, J.; Xie, J.; Parniak, M.A.; Sarafianos, S.G.; et al. 6-Arylthio-3-hydroxypyrimidine-2,4-diones potently inhibited HIV reverse transcriptase-associated RNase H with antiviral activity. Eur. J. Med. Chem. 2018, 156, 652–665. [Google Scholar] [CrossRef]
- Boyer, P.L.; Smith, S.J.; Zhao, X.Z.; Das, K.; Gruber, K.; Arnold, E.; Burke, T.R.; Hughes, S.H. Developing and evaluating inhibitors against the RNase H active site of HIV-1 reverse transcriptase. J. Virol. 2018, 92, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velthuisen, E.J.; Johns, B.A.; Gerondelis, P.; Chen, Y.; Li, M.; Mou, K.; Zhang, W.; Seal, J.W.; Hightower, K.E.; Miranda, S.R.; et al. Pyridopyrimidinone inhibitors of HIV-1 RNase H. Eur. J. Med. Chem. 2014, 83, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.; Di Leva, F.S.; Thierry, S.; Pescatori, L.; Cuzzucoli Crucitti, G.; Subra, F.; Delelis, O.; Esposito, F.; Rigogliuso, G.; Costi, R.; et al. Identification of highly conserved residues involved in inhibition of HIV-1 RNase H function by diketo acid derivatives. Antimicrob. Agents Chemother. 2014, 58, 6101–6110. [Google Scholar] [CrossRef] [Green Version]
- Poongavanam, V.; Corona, A.; Steinmann, C.; Scipione, L.; Grandi, N.; Pandolfi, F.; Di Santo, R.; Costi, R.; Esposito, F.; Tramontano, E.; et al. Structure-guided approach identifies a novel class of HIV-1 ribonuclease H inhibitors: Binding mode insights through magnesium complexation and site-directed mutagenesis studies. Medchemcomm 2018, 9, 562–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enzo, T.; Yung-chi, C. HIV-1 reverse transcriptase inhibition by a dipyridodiazepinone derivative: BI-RG-587. Biochem. Pharmacol. 1992, 43, 1371–1376. [Google Scholar] [CrossRef]
- Vitoria, M.; Rangaraj, A.; Ford, N.; Doherty, M. Current and future priorities for the development of optimal HIV drugs. Curr. Opin. HIV AIDS 2019, 14, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Wensing, A.M.; Calvez, V.; Ceccherini-Silberstein, F.; Charpentier, C.; Günthard, H.F.; Paredes, R.; Shafer, R.W.; Richman, D. 2019 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 2019, 27, 111–121. [Google Scholar]
- Divita, G.; Restle, T.; Goody, R.S.; Chermann, J.C.; Baillon, J.G. Inhibition of human immunodeficiency virus type 1 reverse transcriptase dimerization using synthetic peptides derived from the connection domain. J. Biol. Chem. 1994, 269, 13080–13083. [Google Scholar]
- Sánchez-Murcia, P.A.; de Castro, S.; García-Aparicio, C.; Jiménez, M.A.; Corona, A.; Tramontano, E.; Sluis-Cremer, N.; Menéndez-Arias, L.; Velázquez, S.; Gago, F.; et al. Peptides mimicking the β7/β8 loop of HIV-1 reverse transcriptase p51 as “hotspot-targeted” dimerization inhibitors. ACS Med. Chem. Lett. 2020. [Google Scholar] [CrossRef]
- Camarasa, M.-J.; Velázquez, S.; San-Félix, A.; Pérez-Pérez, M.-J.; Gago, F. Dimerization inhibitors of HIV-1 reverse transcriptase, protease and integrase: A single mode of inhibition for the three HIV enzymes? Antiviral Res. 2006, 71, 260–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tintori, C.; Corona, A.; Esposito, F.; Brai, A.; Grandi, N.; Ceresola, E.R.; Clementi, M.; Canducci, F.; Tramontano, E.; Botta, M.; et al. Inhibition of HIV-1 Reverse Transcriptase Dimerization by Small Molecules. ChemBioChem 2016, 17, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Wendeler, M.; Rausch, J.W.; Beilhartz, G.; Gotte, M.; O’Keefe, B.R.; Bermingham, A.; Beutler, J.A.; Liu, S.; Zhuang, X.; et al. Structure-activity analysis of vinylogous urea inhibitors of human immunodeficiency virus-encoded ribonuclease H. Antimicrob. Agents Chemother. 2010, 54, 3913–3921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himmel, D.M.; Sarafianos, S.G.; Dharmasena, S.; Hossain, M.M.; McCoy-Simandle, K.; Ilina, T.; Clark, A.D.; Knight, J.L.; Julias, J.G.; Clark, P.K.; et al. HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl naphthyl hydrazone bound at a novel site. ACS Chem. Biol. 2006, 1, 702–712. [Google Scholar] [CrossRef] [Green Version]
- Christen, M.T.; Menon, L.; Myshakina, N.S.; Ahn, J.; Parniak, M.A.; Ishima, R. Structural basis of the allosteric inhibitor interaction on the HIV-1 reverse transcriptase RNase H domain. Chem. Biol. Drug Des. 2012, 80, 706–716. [Google Scholar] [CrossRef] [Green Version]
- Gong, Q.; Menon, L.; Ilina, T.; Miller, L.G.; Ahn, J.; Parniak, M.A.; Ishima, R. Interaction of HIV-1 reverse transcriptase ribonuclease H with an acylhydrazone inhibitor. Chem. Biol. Drug Des. 2011, 77, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Distinto, S.; Maccioni, E.; Meleddu, R.; Corona, A.; Alcaro, S.; Tramontano, E. Molecular aspects of the RT/drug interactions. Perspective of dual inhibitors. Curr. Pharm. Des. 2013, 19, 1850–1859. [Google Scholar] [CrossRef]
- Meleddu, R.; Cannas, V.; Distinto, S.; Sarais, G.; Del Vecchio, C.; Esposito, F.; Bianco, G.; Corona, A.; Cottiglia, F.; Alcaro, S.; et al. Design, synthesis, and biological evaluation of 1,3-diarylpropenones as dual inhibitors of HIV-1 reverse transcriptase. ChemMedChem 2014, 9, 1869–1879. [Google Scholar] [CrossRef]
- Corona, A.; Onnis, V.; Deplano, A.; Bianco, G.; Demurtas, M.; Distinto, S.; Cheng, Y.C.; Alcaro, S.; Esposito, F.; Tramontano, E. Design, synthesis and antiviral evaluation of novel heteroarylcarbothioamide derivatives as dual inhibitors of HIV-1 Reverse transcriptase-associated RNase H and RDDP functions. Pathog. Dis. 2017, 75, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Meleddu, R.; Distinto, S.; Corona, A.; Tramontano, E.; Bianco, G.; Melis, C.; Cottiglia, F.; Maccioni, E.; Meleddu, R.; Distinto, S.; et al. Isatin thiazoline hybrids as dual inhibitors of HIV-1 reverse transcriptase. J. Enzyme Inhib. Med. Chem. 2017, 32, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Corona, A.; Meleddu, R.; Esposito, F.; Distinto, S.; Bianco, G.; Masaoka, T.; Maccioni, E.; Menéndez-Arias, L.; Alcaro, S.; Le Grice, S.F.J.; et al. Ribonuclease H/DNA Polymerase HIV-1 Reverse Transcriptase Dual Inhibitor: Mechanistic Studies on the Allosteric Mode of Action of Isatin-Based Compound RMNC6. PLoS ONE 2016, 11, e0147225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Distinto, S.; Esposito, F.; Kirchmair, J.; Cardia, M.C.; Gaspari, M.; MacCioni, E.; Alcaro, S.; Markt, P.; Wolber, G.; Zinzula, L.; et al. Identification of HIV-1 reverse transcriptase dual inhibitors by a combined shape-, 2D-fingerprint- and pharmacophore-based virtual screening approach. Eur. J. Med. Chem. 2012, 50, 216–229. [Google Scholar] [CrossRef] [PubMed]
- de Castro, S.; Camarasa, M.J. Polypharmacology in HIV inhibition: Can a drug with simultaneous action against two relevant targets be an alternative to combination therapy? Eur. J. Med. Chem. 2018, 150, 206–227. [Google Scholar] [CrossRef] [PubMed]
- Judge, V.; Narasimhan, B.; Ahuja, M.; Sriram, D.; Yogeeswari, P.; De Clercq, E.; Pannecouque, C.; Balzarini, J. Synthesis, antimycobacterial, antiviral, antimicrobial activities, and QSAR studies of isonicotinic acid-1-(substituted phenyl)-ethylidene/cycloheptylidene hydrazides. Med. Chem. Res. 2012, 21, 1935–1952. [Google Scholar] [CrossRef]
- Narang, R.; Narasimhan, B.; Sharma, S.; Sriram, D.; Yogeeswari, P.; De Clercq, E.; Pannecouque, C.; Balzarini, J. Synthesis, antimycobacterial, antiviral, antimicrobial activities, and QSAR studies of nicotinic acid benzylidene hydrazide derivatives. Med. Chem. Res. 2012, 21, 1557–1576. [Google Scholar] [CrossRef]
- Cocco, M.T.; Congiu, C.; Onnis, V.; Morelli, M.; Felipo, V.; Cauli, O. Synthesis of new 2-arylamino-6-trifluoromethylpyridine-3-carboxylic acid derivatives and investigation of their analgesic activity. Bioorg. Med. Chem. 2004, 12, 4169–4177. [Google Scholar] [CrossRef]
- Onnis, V.; Cocco, M.T.; Lilliu, V.; Congiu, C. Synthesis and evaluation of antitumoral activity of ester and amide derivatives of 2-arylamino-6-trifluoromethyl-3-pyridinecarboxylic acids. Bioorganic Med. Chem. 2008, 16, 2367–2378. [Google Scholar] [CrossRef]
- Himmel, D.M.; Maegley, K.A.; Pauly, T.A.; Bauman, J.D.; Das, K.; Dharia, C.; Clark, A.D.; Ryan, K.; Hickey, M.J.; Love, R.A.; et al. Structure of HIV-1 reverse transcriptase with the inhibitor beta-Thujaplicinol bound at the RNase H active site. Structure 2009, 17, 1625–1635. [Google Scholar] [CrossRef] [Green Version]
- Alcaro, S.; Artese, A.; Ceccherini-Silberstein, F.; Chiarella, V.; Dimonte, S.; Ortuso, F.; Perno, C.F. Computational analysis of Human Immunodeficiency Virus (HIV) Type-1 reverse transcriptase crystallographic models based on significant conserved residues found in Highly Active Antiretroviral Therapy (HAART)-treated patients. Curr. Med. Chem. 2010, 17, 290–308. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.U. Polypharmacology—Foe or friend? J. Med. Chem. 2013, 56, 8955–8971. [Google Scholar] [CrossRef]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Gao, P.; Dong, G.; Zhang, X.; Cheng, X.; Ding, X.; Wang, X.; Daelemans, D.; De Clercq, E.; Pannecouque, C.; et al. 5-Hydroxypyrido[2,3-b]pyrazin-6(5H)-one derivatives as novel dual inhibitors of HIV-1 reverse transcriptase-associated ribonuclease H and integrase. Eur. J. Med. Chem. 2018, 155, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Cuzzucoli Crucitti, G.; Métifiot, M.; Pescatori, L.; Messore, A.; Madia, V.N.; Pupo, G.; Saccoliti, F.; Scipione, L.; Tortorella, S.; Esposito, F.; et al. Structure-activity relationship of pyrrolyl diketo acid derivatives as dual inhibitors of HIV-1 integrase and reverse transcriptase ribonuclease H domain. J. Med. Chem. 2015, 58, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Costi, R.; Métifiot, M.; Chung, S.; Cuzzucoli Crucitti, G.; Maddali, K.; Pescatori, L.; Messore, A.; Madia, V.N.; Pupo, G.; Scipione, L.; et al. Basic Quinolinonyl Diketo Acid Derivatives as Inhibitors of HIV Integrase and their Activity against RNase H Function of Reverse Transcriptase. J. Med. Chem. 2014, 57, 3223–3234. [Google Scholar] [CrossRef]
- Carcelli, M.; Rogolino, D.; Sechi, M.; Rispoli, G.; Fisicaro, E.; Compari, C.; Grandi, N.; Corona, A.; Tramontano, E.; Pannecouque, C.; et al. Antiretroviral activity of metal-chelating HIV-1 integrase inhibitors. Eur. J. Med. Chem. 2014, 83, 594–600. [Google Scholar] [CrossRef]
- Gao, P.; Cheng, X.; Sun, L.; Song, S.; Álvarez, M.; Luczkowiak, J.; Pannecouque, C.; De Clercq, E.; Menéndez-Arias, L.; Zhan, P.; et al. Design, synthesis and biological evaluation of 3-hydroxyquinazoline-2,4(1H,3H)-diones as dual inhibitors of HIV-1 reverse transcriptase-associated RNase H and integrase. Bioorganic Med. Chem. 2019, 4, 1–10. [Google Scholar] [CrossRef]
- Esposito, F.; Sechi, M.; Pala, N.; Sanna, A.; Koneru, P.C.; Kvaratskhelia, M.; Naesens, L.; Corona, A.; Grandi, N.; di Santo, R.; et al. Discovery of dihydroxyindole-2-carboxylic acid derivatives as dual allosteric HIV-1 Integrase and reverse transcriptase associated ribonuclease H inhibitors. Antivir. Res. 2020, 174, 104671. [Google Scholar] [CrossRef]
- Martini, R.; Esposito, F.; Corona, A.; Ferrarese, R.; Ceresola, E.R.; Visconti, L.; Tintori, C.; Barbieri, A.; Calcaterra, A.; Iovine, V.; et al. Natural Product Kuwanon-L Inhibits HIV-1 Replication through Multiple Target Binding. ChemBioChem 2017, 18, 374–377. [Google Scholar] [CrossRef]
- Massari, S.; Corona, A.; Distinto, S.; Desantis, J.; Caredda, A.; Sabatini, S.; Manfroni, G.; Felicetti, T.; Cecchetti, V.; Pannecouque, C.; et al. From cycloheptathiophene-3-carboxamide to oxazinone-based derivatives as allosteric HIV-1 ribonuclease H inhibitors. J. Enzyme Inhib. Med. Chem. 2019, 34, 55–74. [Google Scholar] [CrossRef] [Green Version]
- Esposito, F.; Tramontano, E. Past and future. Current drugs targeting HIV-1 integrase and reverse transcriptase-associated ribonuclease H activity: Single and dual active site inhibitors. Antivir. Chem. Chemother. 2013, 23, 129–144. [Google Scholar] [CrossRef] [Green Version]
- Bahar, I.; Erman, B.; Jernigan, R.L.; Atilgan, A.R.; Covell, D.G. Collective motions in HIV-1 reverse transcriptase: Examination of flexibility and enzyme function. J. Mol. Biol. 1999, 285, 1023–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meleddu, R.; Distinto, S.; Corona, A.; Bianco, G.; Cannas, V.; Esposito, F.; Artese, A.; Alcaro, S.; Matyus, P.; Bogdan, D.; et al. (3Z)-3-(2-[4-(aryl)-1,3-thiazol-2-yl]hydrazin-1-ylidene)-2,3-dihydro- 1H -indol-2-one derivatives as dual inhibitors of HIV-1 reverse transcriptase. Eur. J. Med. Chem. 2015, 93, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Carli, I.; Del Vecchio, C.; Xu, L.; Corona, A.; Grandi, N.; Piano, D.; Maccioni, E.; Distinto, S.; Parolin, C.; et al. Sennoside A, derived from the traditional chinese medicine plant Rheum L., is a new dual HIV-1 inhibitor effective on HIV-1 replication. Phytomedicine 2016, 23, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Pala, N.; Esposito, F.; Rogolino, D.; Carcelli, M.; Sanna, V.; Palomba, M.; Naesens, L.; Corona, A.; Grandi, N.; Tramontano, E.; et al. Inhibitory effect of 2,3,5,6-tetrafluoro-4-[4-(Aryl)-1H-1,2,3-triazol-1-yl]benzenesulfonamide derivatives on HIV reverse transcriptase associated RNase H activities. Int. J. Mol. Sci. 2016, 17, 1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, G.; Rocca, R.; Corona, A.; Grandi, N.; Moraca, F.; Romeo, I.; Talarico, C.; Giovanna, M.; Alessandra, F.; Ortuso, F.; et al. Novel natural non-nucleoside inhibitors of HIV-1 reverse transcriptase identified by shape- and structure-based virtual screening techniques. Eur. J. Med. Chem. 2019, 161, 1–10. [Google Scholar] [CrossRef]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are available from V.O. the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R | R’ | RNase H IC50 (µM) a |
|---|---|---|---|
| 6 | H | >100 (87%) b | |
| 9 | OEt | 24.0 ± 1.7 | |
| 11 | 2-Cl-phenyl | >100 (92%) | |
| 12 | 3-Cl-phenyl | >100 (98%) | |
| 13 | 4-Cl-phenyl | 9.3 ± 2.5 | |
| 14 | 2,4-Cl2-phenyl | >100 (98%) | |
| 15 | 2,4,6-Cl3-phenyl | 31.8 ± 3.5 | |
| 16 | 2-OH-phenyl | 16.2 ± 6.6 | |
| 17 | 3-OH-phenyl | 26.6 ± 5.6 | |
| 18 | 4-OH-phenyl | 20.0 ± 1.3 | |
| 19 | 3-OCH3-phenyl | >100 (80%) | |
| 20 | 3,4-(OCH3)2-phenyl | 21 ± 11 | |
| 21 | 3,5-(OCH3)2-phenyl | 14.7 ± 1.4 | |
| 22 | 3,4,5-(OCH3)3-phenyl | 17.3 ± 1.3 | |
| 23 | 4-OCH3-phenyl | 11.6 ± 4.3 | |
| 24 | 4-SCH3-phenyl | 9.6 ± 0.8 | |
| 25 | OH | 0.77 ± 0.06 | |
| 26 | CH2COOEt | >100 (100%) | |
| 27 | 3-pyridyl | 25.0 ± 4.9 | |
| 28 | 4-SCH3-phenyl | 5.9 ± 0.9 | |
| 29 | 3-CH3-phenyl | 8.9 ± 0.1 | |
| 30 | 3-Cl-phenyl | 4.0 ± 2.2 | |
| 31 | 2,4-Cl2-phenyl | 8.8 ± 0.5 | |
| 32 | 3-Cl, 4-OCH3 | >100 (100%) | |
| 33 | 3-OCH3-phenyl | >100 (54%) | |
| 34 | 3,5-(OCH3)2-phenyl | 17.5 ± 3.9 | |
| 35 | 3,4-(OCH3)2-phenyl | 22.4 ± 4.0 | |
| 36 | 3,4,5-(OCH3)3-phenyl | 19.4 ± 1.5 | |
| 37 | 4-CF3-phenyl | 7.8 ± 1.2 | |
| 38 | 2-Cl-benzyl | 19.4 ± 1.1 | |
| 39 | 2-OCH3-benzyl | 18.7 ± 1.7 | |
| 40 | 4-Cl-benzyl | 6.4 ± 0.5 | |
| 41 | 4-F-benzyl | 23.3 ± 3.1 | |
| 42 | 4-CF3-benzyl | 21.9 ± 0.2 | |
| 43 | 4-OCH3-benzyl | 30.4 ± 3.9 | |
| 44 | 4-CH3-benzyl | 21.3 ± 0.5 | |
| 45 | 2,4-Cl2-benzyl | 19.3 ± 0.6 | |
| 46 | 3,4-Cl2-benzyl | 25.6 ± 3.4 | |
| 47 | 3-OCH3, 4-OH-benzyl | 18.2 ± 1.7 | |
| 48 | 4-phenylpiperazino | >100 (100%) | |
| 49 | 4-(3-Cl-phenyl)piperazino | 18.0 ± 5.7 | |
| 50 | 4-(4-F-phenyl)piperazino | >100 (100%) | |
| 51 | 4-(4-OCH3-phenyl)piperazino | 20.8 ± 2.5 | |
| 52 | 4-(3,4-Cl2-phenyl)piperazino | 7.5 ± 2.0 | |
| RDS1759 | - | - | 8.5 ± 0.5 |
| BTP | - | - | 0.19 ± 0.03 |
| Compoud | HIV-1 a EC50 (μM) | b CC50 (μM) | SI d | |
|---|---|---|---|---|
| TZMbl Cells | TZMbl | CEM Cells | ||
| 9 | >10 | >50 (75%) c | >50 (100%) | - |
| 13 | 10 | 36 ± 1 | >50 (70%) | 3.6 |
| 16 | >10 | 12.2 ± 2.1 | >12.5 (100%) | - |
| 17 | >50 | >50 (62%) | >50 (100%) | - |
| 21 | 5 ± 0.8 | >50 (59%) | >50 (100%) | >10 |
| 23 | >10 | >50 (70%) | >50 (100%) | - |
| 24 | >10 | 50 | >50 (100%) | - |
| 25 | >10 | 14 ± 2.8 | 35 | - |
| 27 | >5.8 | 5.8 ± 0.8 | 16.2 | - |
| 29 | 2.5 ± 0.4 | 3.9 ± 0.9 | 6.3 | 1.6 |
| 30 | >3.9 | 3.9 ± 0.9 | 8.4 | - |
| 31 | >4.7 | 4.7 ± 1.7 | 7.8 | - |
| 34 | 2 ± 0.3 | 5.3 ± 0.6 | 10.4 | 2.6 |
| 36 | 1.8 ± 0.6 | 2.2 ± 0.4 | 10.2 | 1.2 |
| 37 | >2.2 | 2.2 ± 0.2 | 4.3 | - |
| 38 | >5.7 | 5.7 ± 0.7 | 12.5 | - |
| 39 | >10 | 14.8 ± 3.1 | >12.5 (55%) | - |
| 40 | >4.7 | 4.7 ± 0.4 | 7.75 | - |
| 41 | >6 | 6 ± 0.6 | 11.6 | - |
| 42 | >3.6 | 3.6 ± 1.2 | 5 | - |
| 43 | >10 | 13.2 ± 1.3 | 20 | - |
| 44 | >1.1 | 1.1 ± 0 | 3.5 | - |
| 45 | >10 | 5 ± 0.6 | 9.15 | - |
| 46 | >6.3 | 6.3 ± 1.3 | 10.6 | - |
| 47 | >10 | 15 ± 3 | 17.4 | - |
| 49 | 10 ± 1.5 | 27 ± 3 | >50 (82%) | 2.7 |
| 51 | >10 | 27.7 ± 12.8 | >50 (81%) | - |
| 52 | 10 ± 1.2 | 15.7 ± 5.1 | >50 (76%) | 1.6 |
| EFV | 0.15 ± 0.02 | >50 (100%) | >50 (100%) | 357 |
| 21 | 49 | EFV | ||||
|---|---|---|---|---|---|---|
| RT | IC50 (µM) a | Foldc | IC50 (µM) a | Fold | IC50 (nM) a | Fold |
| WT | 16.1 ± 1.2 | 22.9 ± 1.4 | 23 ± 4.1 | 1 | ||
| K103N | 16.5 ± 4.8 | 1.0 | 68.6 ± 3.0 | 2.5 | 176 ± 25 | 7.6 |
| Y181C | 22.5 ± 5.3 | 1.4 | 46.4 ± 2.4 | 2.0 | 49.7 ± 9.1 | 2.2 |
| V108A | 82.8 ± 6.3 | 5.1 | >100 (54%) b | > 4.3 | 21.3 ± 3.6 | 0.9 |
| Q475A | 17.0 ± 4.5 | 1.0 | 57.7 ± 2.1 | 2.5 | 34.5 ± 2.3 | 1.5 |
| A502F | 8.0 ± 0.7 | 0.5 | 27.0 ± 1.7 | 1.2 | 24.7 ± 2.4 | 1.1 |
| 21 | 49 | BTP | ||||
|---|---|---|---|---|---|---|
| RT | IC50 (µM) a | Fold | IC50 (µM) | Fold | IC50 (µM) | Fold |
| WT | 14.7 ± 1.4 | 18.0 ± 5.7 | 0.19 ± 0.03 | 1 | ||
| K103N | 53.9 ± 2.2 | 3.7 | 41.0 ± 2.2 | 2.3 | 0.22 ± 0.08 | 1.2 |
| Y181C | 11.9 ± 1.4 | 0.8 | 55.4 ± 12.6 | 3.1 | 0.23 ± 0.05 | 1.2 |
| V108A | >100 (100%) b | >6.8 | >100 (100%) | >5.5 | 0.34 ± 0.4 | 1.8 |
| Q475A | >100 (100%) | >6.8 | >100 (100%) | >5.5 | 0.19 ± 0.03 | 1.0 |
| A502F | >100 (55%) | >6.8 | 47.7 ± 5.4 | 2.6 | 0.17 ± 0.03 | 0.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corona, A.; Onnis, V.; Del Vecchio, C.; Esposito, F.; Cheng, Y.-C.; Tramontano, E. 2-(Arylamino)-6-(trifluoromethyl)nicotinic Acid Derivatives: New HIV-1 RT Dual Inhibitors Active on Viral Replication. Molecules 2020, 25, 1338. https://doi.org/10.3390/molecules25061338
Corona A, Onnis V, Del Vecchio C, Esposito F, Cheng Y-C, Tramontano E. 2-(Arylamino)-6-(trifluoromethyl)nicotinic Acid Derivatives: New HIV-1 RT Dual Inhibitors Active on Viral Replication. Molecules. 2020; 25(6):1338. https://doi.org/10.3390/molecules25061338
Chicago/Turabian StyleCorona, Angela, Valentina Onnis, Claudia Del Vecchio, Francesca Esposito, Yung-Chi Cheng, and Enzo Tramontano. 2020. "2-(Arylamino)-6-(trifluoromethyl)nicotinic Acid Derivatives: New HIV-1 RT Dual Inhibitors Active on Viral Replication" Molecules 25, no. 6: 1338. https://doi.org/10.3390/molecules25061338
APA StyleCorona, A., Onnis, V., Del Vecchio, C., Esposito, F., Cheng, Y. -C., & Tramontano, E. (2020). 2-(Arylamino)-6-(trifluoromethyl)nicotinic Acid Derivatives: New HIV-1 RT Dual Inhibitors Active on Viral Replication. Molecules, 25(6), 1338. https://doi.org/10.3390/molecules25061338