Mass Spectrometry Advances and Perspectives for the Characterization of Emerging Adoptive Cell Therapies

Abstract
:1. Introduction
2. CAR T-Cell Therapy Manufacturing and State-of-The-Art Analytics
2.1. Manufacturing of CAR-T
2.2. Overview of Mass Spectrometry-Based Proteomics
2.3. Mass Spectrometry to Decipher the Mechanism of Action of CAR-T Therapies
3. Advances in Mass Spectrometry that Will Benefit CAR T-Cell Therapy Characterization
3.1. Advances in Sample Preparation
3.2. Advances in Peptide Separation
3.3. A New Venue for MS-Based Proteomics: Single-cell Analysis
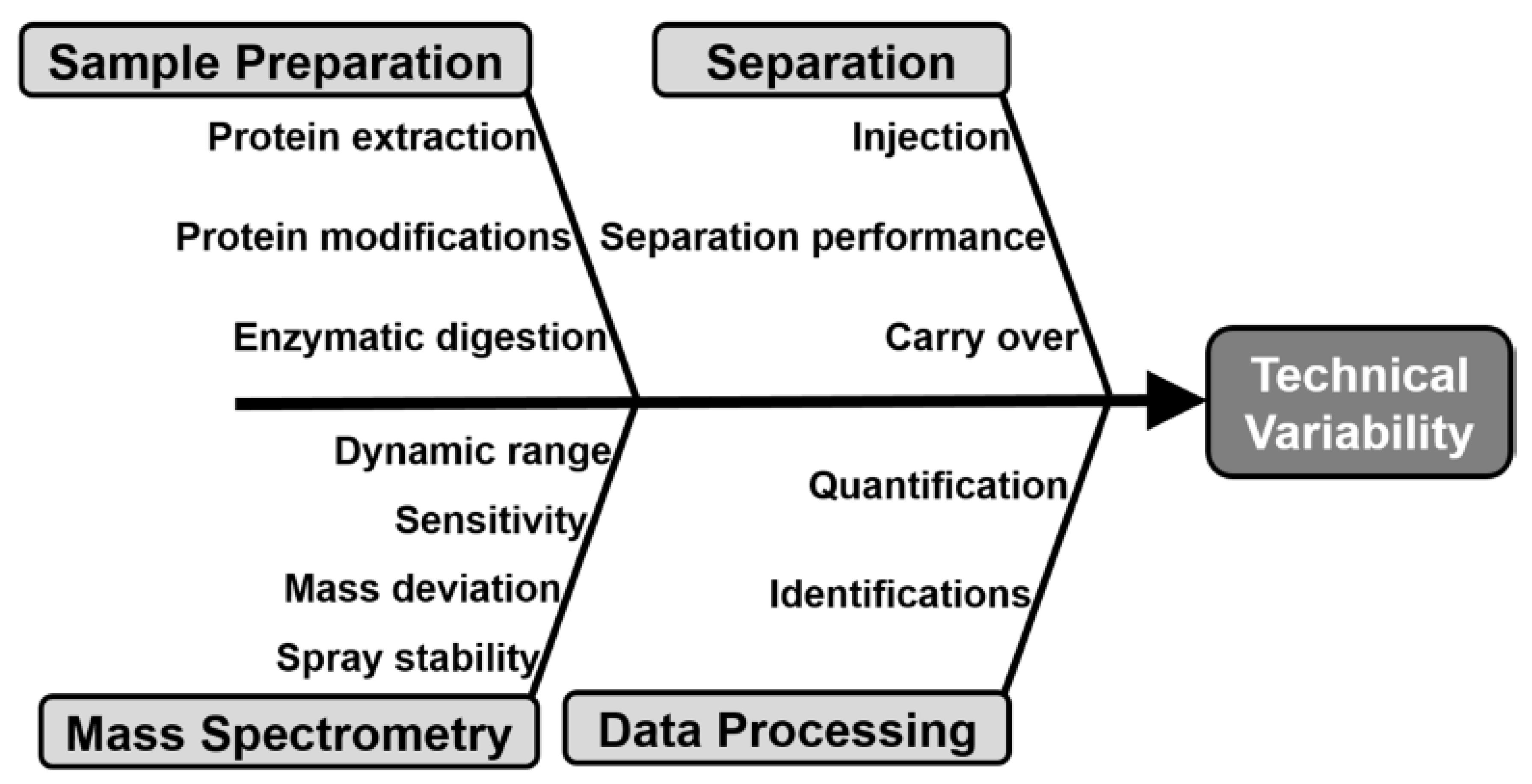
4. Implementation of Measurement Controls
5. Conclusions
Disclaimer
Funding
Acknowledgments
Conflicts of Interest
References
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Peshkin, L.; Wühr, M.; Pearl, E.; Haas, W.; Robert, M.F.; John, C.G.; Allon, M.K.; Horb, M.; Steven, P.G.; Marc, W.K. On the relationship of protein and mRNA dynamics in vertebrate embryonic development. Dev. Cell 2015, 35, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Yates, J.R. The revolution and evolution of shotgun proteomics for large-scale proteome analysis. J. Am. Chem. Soc. 2013, 135, 1629–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, B.; Achour, B.; Artursson, P.; Hop, C.; Lai, Y.R.; Smith, P.C.; Barber, J.; Wisniewski, J.R.; Spellman, D.; Uchida, Y.; et al. Toward a consensus on applying quantitative liquid chromatography-tandem mass spectrometry proteomics in translational pharmacology research: A white paper. Clin. Pharmacol. Ther. 2019, 106, 525–543. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, A.; Coish, J.M.; Yeung, J.; Muselius, B.; Gadjeva, M.; Macneil, A.J.; Geddes-Mcalister, J. Decoding communication patterns of the innate immune system by quantitative proteomics. J. Leukocyte Biol. 2019, 106, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.; Cao, L.L.; Lin, J.T.; Hung, N.; Ritz, A.; Yu, K.B.; Jianu, R.; Ulin, S.P.; Raphael, B.J.; Laidlaw, D.H. A new approach for quantitative phosphoproteomic dissection of signaling pathways applied to T-cell receptor activation. Mol. Cell. Proteomics 2009, 8, 2418–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruperez, P.; Gago-Martinez, A.; Burlingame, A.L.; Oses-Prieto, J.A. Quantitative phosphoproteomic analysis reveals a role for serine and threonine kinases in the cytoskeletal reorganization in early T-cell receptor activation in Human primary T-cells. Mol. Cell. Proteomics 2012, 11, 171–186. [Google Scholar] [CrossRef] [Green Version]
- Navarro, M.N.; Goebel, J.; Feijoo-Carnero, C.; Morrice, N.; Cantrell, D.A. Phosphoproteomic analysis reveals an intrinsic pathway for the regulation of histone deacetylase 7 that controls the function of cytotoxic T lymphocytes. Nat. Immunol. 2011, 12, 352–361. [Google Scholar] [CrossRef] [PubMed]
- McBride, D.A.; Kerr, M.D.; Wai, S.L.; Shah, N.J. Applications of molecular engineering in T-cell-based immunotherapies. Wiley Interdiscip. Rev.-Nanomed. Nanobiotechnol. 2019, 11, 25. [Google Scholar] [CrossRef]
- Harris, D.T.; Kranz, D.M. Adoptive T-cell therapies: A comparison of T-cell receptors and chimeric antigen receptors. Trends Pharmacol. Sci. 2016, 37, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Harris, D.T.; Hager, M.V.; Smith, S.N.; Cai, Q.; Stone, J.D.; Kruger, P.; Lever, M.; Dushek, O.; Schmitt, T.M.; Greenberg, P.D.; et al. Comparison of T-cell activities mediated by human TCRs and CARs that use the same recognition domains. J. Immunol. 2018, 200, 1088–1100. [Google Scholar] [CrossRef]
- Calmes-Miller, J. FDA approves second CAR T-cell therapy. Cancer Discov. 2018, 8, 5–6. [Google Scholar]
- Rivière, I.; Roy, K. Perspectives on manufacturing of high-quality cell therapies. Mol.Ther. 2017, 25, 1067–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Rivière, I. Clinical manufacturing of CAR T cells: Foundation of a promising therapy. Mol. Ther. Oncolytics 2016, 3, 16015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebert, A.S.; Richards, A.L.; Bailey, D.J.; Ulbrich, A.; Coughlin, E.E.; Westphall, M.S.; Coon, J.J. The one hour yeast proteome. Mol. Cell. Proteomics 2014, 13, 339–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.L.; Bertke, M.M.; Champion, M.M.; Zhu, G.J.; Huber, P.W.; Dovichi, N.J. Quantitative proteomics of Xenopus laevis embryos: Expression kinetics of nearly 4000 proteins during early development. Sci. Rep. 2014, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Wuehr, M.; Freeman, R.M.; Presler, M.; Horb, M.E.; Peshkin, L.; Gygi, S.P.; Kirschner, M.W. Deep proteomics of the Xenopus laevis egg using an mRNA-derived reference database. Curr. Biol. 2014, 24, 1467–1475. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; Schmitt, S.; Bergner, C.G.; Tyanova, S.; Kannaiyan, N.; Manrique-Hoyos, N.; Kongi, K.; Cantuti, L.; Hanisch, U.K.; Philips, M.A.; et al. Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 2015, 18, 1819–1831. [Google Scholar] [CrossRef]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 10. [Google Scholar] [CrossRef]
- Manes, N.P.; Nita-Lazar, A. Application of targeted mass spectrometry in bottom-up proteomics for systems biology research. J. Proteomics 2018, 189, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Vidova, V.; Spacil, Z. A review on mass spectrometry-based quantitative proteomics: Targeted and data independent acquisition. Anal. Chim. Acta 2017, 964, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Aslanian, A.; Yates, J.R. Mass spectrometry for proteomics. Curr. Opin. Chem. Biol. 2008, 12, 483–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.-C.; Yates, J.R. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moresco, J.J.; Carvalho, P.C.; Yates, J.R. Identifying components of protein complexes in C. elegans using co-immunoprecipitation and mass spectrometry. J. Proteomics 2010, 73, 2198–2204. [Google Scholar] [CrossRef] [Green Version]
- Paul, F.E.; Hosp, F.; Selbach, M. Analyzing protein-protein interactions by quantitative mass spectrometry. Methods 2011, 54, 387–395. [Google Scholar] [CrossRef]
- Rudashevskaya, E.L.; Sickmann, A.; Markoutsa, S. Global profiling of protein complexes: Current approaches and their perspective in biomedical research. Expert Rev. Proteomics 2016, 13, 951–964. [Google Scholar] [CrossRef]
- Sun, L.L.; Zhu, G.J.; Yan, X.J.; Zhang, Z.B.; Wojcik, R.; Champion, M.M.; Dovichi, N.J. Capillary zone electrophoresis for bottom-up analysis of complex proteomes. Proteomics 2016, 16, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Gomes, F.P.; Yates, J.R., III. Recent trends of capillary electrophoresis-mass spectrometry in proteomics research. Mass Spectrom. Rev. 2019, 38, 445–460. [Google Scholar] [CrossRef]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.; Aebersold, R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, 23. [Google Scholar] [CrossRef]
- Levin, Y.; Hradetzky, E.; Bahn, S. Quantification of proteins using data-independent analysis (MSE) in simple and complex samples: A systematic evaluation. Proteomics 2011, 11, 3273–3287. [Google Scholar] [CrossRef] [PubMed]
- Egertson, J.D.; Kuehn, A.; Merrihew, G.E.; Bateman, N.W.; MacLean, B.X.; Ting, Y.S.; Canterbury, J.D.; Marsh, D.M.; Kellmann, M.; Zabrouskov, V.; et al. Multiplexed MS/MS for improved data-independent acquisition. Nat. Methods 2013, 10, 744. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, Y.; Watanabe, E.; Umeyama, T.; Nakajima, D.; Hattori, M.; Honda, K.; Ohara, O. Optimization of data-independent acquisition mass spectrometry for deep and highly sensitive proteomic analysis. Int. J. Mol. Sci. 2019, 20, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappireddi, N.; Martin, L.; Wuhr, M. A review on quantitative multiplexed proteomics. Chem. Bio. Chem. 2019, 20, 1210–1224. [Google Scholar] [CrossRef]
- Bonneil, E.; Pfammatter, S.; Thibault, P. Enhancement of mass spectrometry performance for proteomic analyses using high-field asymmetric waveform ion mobility spectrometry (FAIMS). J. Mass Spectrom. 2015, 50, 1181–1195. [Google Scholar] [CrossRef]
- Chouinard, C.D.; Nagy, G.; Webb, I.K.; Shi, T.J.; Baker, E.S.; Prost, S.A.; Liu, T.; Ibrahim, Y.M.; Smith, R.D. Improved sensitivity and separations for phosphopeptides using online liquid chromotography coupled with structures for lossless ion manipulations ion mobility-mass spectrometry. Anal. Chem. 2018, 90, 10889–10896. [Google Scholar] [CrossRef]
- Distler, U.; Kuharev, J.; Navarro, P.; Tenzer, S. Label-free quantification in ion mobility-enhanced data-independent acquisition proteomics. Nat. Protoc. 2016, 11, 795–812. [Google Scholar] [CrossRef]
- Fouque, K.J.D.; Fernandez-Lima, F. Recent advances in biological separations using trapped ion mobility spectrometry–mass spectrometry. Trac-Trends Anal. Chem. 2019, 116, 308–315. [Google Scholar] [CrossRef]
- Meier, F.; Beck, S.; Grassl, N.; Lubeck, M.; Park, M.A.; Raether, O.; Mann, M. Parallel accumulation-serial fragmentation (PASEF): Multiplying sequencing speed and sensitivity by synchronized scans in a trapped ion mobility device. J. Proteome Res. 2015, 14, 5378–5387. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online parallel accumulation serial fragmentation (PASEF) with a novel trapped on mobility mass spectrometer. Mol. Cell. Proteomics 2018, 17, 2534–2545. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Apweiler, R.; Alpi, E.; Antunes, R.; Ar-Ganiska, J.; Bely, B.; Bingley, M.; et al. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Menschaert, G.; Van Criekinge, W.; Notelaers, T.; Koch, A.; Crappe, J.; Gevaert, K.; Van Damme, P. Deep proteome coverage based on ribosome profiling aids mass spectrometry-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Mol. Cell. Proteomics 2013, 12, 1780–1790. [Google Scholar] [CrossRef] [Green Version]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An approach to correlate tandem mass-spectral data of peptides with amonio acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef] [Green Version]
- Perkins, D.N.; Pappin, D.J.C.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Shilov, I.V.; Seymour, S.L.; Patel, A.A.; Loboda, A.; Tang, W.H.; Keating, S.P.; Hunter, C.L.; Nuwaysir, L.M.; Schaeffer, D.A. The paragon algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteomics 2007, 6, 1638–1655. [Google Scholar] [CrossRef] [Green Version]
- Egertson, J.D.; MacLean, B.; Johnson, R.; Xuan, Y.; MacCoss, M.J. Multiplexed peptide analysis using data-independent acquisition and Skyline. Nat. Protoc. 2015, 10, 887–903. [Google Scholar] [CrossRef] [Green Version]
- Rost, H.L.; Rosenberger, G.; Navarro, P.; Gillet, L.; Miladinovic, S.M.; Schubert, O.T.; Wolskit, W.; Collins, B.C.; Malmstrom, J.; Malmstrom, L.; et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol. 2014, 32, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Tiwary, S.; Levy, R.; Gutenbrunner, P.; Soto, F.S.; Palaniappan, K.K.; Deming, L.; Berndl, M.; Brant, A.; Cimermancic, P.; Cox, J. High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis. Nat. Methods 2019, 16, 519. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, X.; Shen, C.; Lin, Y.; Yang, P.; Qiao, L. In silico spectral libraries by deep learning facilitate data-independent acquisition proteomics. Nat. Commun. 2020, 11, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsou, C.C.; Avtonomov, D.; Larsen, B.; Tucholska, M.; Choi, H.; Gingras, A.C.; Nesvizhskii, A.I. DIA-Umpire: Comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 2015, 12, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindemann, C.; Thomanek, N.; Hundt, F.; Lerari, T.; Meyer, H.E.; Wolters, D.; Marcus, K. Strategies in relative and absolute quantitative mass spectrometry based proteomics. Biol. Chem. 2017, 398, 687–699. [Google Scholar] [CrossRef]
- Arul, A.B.; Robinson, R.A.S. Sample multiplexing strategies in quantitative proteomics. Anal. Chem. 2019, 91, 178–189. [Google Scholar] [CrossRef]
- Neilson, K.A.; Ali, N.A.; Muralidharan, S.; Mirzaei, M.; Mariani, M.; Assadourian, G.; Lee, A.; van Sluyter, S.C.; Haynes, P.A. Less label, more free: Approaches in label-free quantitative mass spectrometry. Proteomics 2011, 11, 535–553. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Hein, M.Y.; Cox, J.; Mann, M. A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol. Cell. Proteomics 2014, 13, 3497–3506. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 1, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef]
- Ross, P.L.; Huang, Y.L.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, F.; Ye, H.; Chen, R.B.; Fu, Q.; Li, L.J. N,N-Dimethyl leucines as novel isobaric tandem mass tags for quantitative proteomics and peptidomics. Anal. Chem. 2010, 82, 2817–2825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadlmeier, M.; Bogena, J.; Wallner, M.; Wuhr, M.; Carell, T. A Sulfoxide-Based Isobaric Labelling Reagent for Accurate Quantitative Mass Spectrometry. Angew. Chem. Int. Ed. 2018, 57, 2958–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minogue, C.E.; Hebert, A.S.; Rensvold, J.W.; Westphall, M.S.; Pagliarini, D.J.; Coon, J.J. Multiplexed quantification for data-independent acquisition. Anal. Chem. 2015, 87, 2570–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuhr, M.; Haas, W.; McAlister, G.C.; Peshkin, L.; Rad, R.; Kirschner, M.W.; Gygi, S.P. Accurate multiplexed proteomics at the MS2 level using the complement reporter ion cluster. Anal. Chem. 2012, 84, 9214–9221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, H.Y.; Muruganujan, A.; Huang, X.S.; Ebert, D.; Mills, C.; Guo, X.Y.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 2015, 162, 425–440. [Google Scholar] [CrossRef] [Green Version]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramello, M.C.; Benzaid, I.; Kuenzi, B.M.; Lienlaf-Moreno, M.; Kandell, W.M.; Santiago, D.N.; Pabon-Saldana, M.; Darville, L.; Fang, B.; Rix, U.; et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci. Signal. 2019, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Weston, L.A.; Bauer, K.M.; Hummon, A.B. Comparison of bottom-up proteomic approaches for LC–MS analysis of complex proteomes. Anal. Methods 2013, 5, 4615–4621. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Ostasiewicz, P.; Mann, M. High recovery FASP applied to the proteomic analysis of microdissected formalin fixed paraffin embedded cancer tissues retrieves known colon cancer markers. J. Proteome Res. 2011, 10, 3040–3049. [Google Scholar] [CrossRef]
- Hughes, C.S.; Foehr, S.; Garfield, D.A.; Furlong, E.E.; Steinmetz, L.M.; Krijgsveld, J. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 2014, 10, 757. [Google Scholar] [CrossRef]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef]
- Zougman, A.; Selby, P.J.; Banks, R.E. Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics 2014, 14, 1006–1010. [Google Scholar] [CrossRef]
- Chen, W.D.; Adhikari, S.; Chen, L.; Lin, L.; Li, H.; Luo, S.S.; Yang, P.Y.; Tian, R.J. 3D-SISPROT: A simple and integrated spintip-based protein digestion and three-dimensional peptide fractionation technology for deep proteome profiling. J. Chromatogr. 2017, 1498, 207–214. [Google Scholar] [CrossRef]
- Myers, S.A.; Rhoads, A.; Cocco, A.R.; Peckner, R.; Haber, A.; Schweitzer, L.D.; Krug, K.; Mani, D.R.; Clauser, K.R.; Rozenblatt-Rosen, O.; et al. Streamlined protocol for deep proteomic profiling of FAC-sorted cells and its application to freshly isolated murine immune cells. Mol. Cell. Proteomics 2019. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef] [PubMed]
- Hailemariam, M.; Eguez, R.V.; Singh, H.; Bekele, S.; Ameni, G.; Pieper, R.; Yu, Y. S-Trap, an ultrafast sample-preparation approach for shotgun proteomics. J. Proteome Res. 2018, 17, 2917–2924. [Google Scholar] [CrossRef]
- Elinger, D.; Gabashvili, A.; Levin, Y. Suspension trapping (S-Trap) is compatible with typical protein extraction buffers and detergents for bottom-up proteomics. J. Proteome Res. 2019, 18, 1441–1445. [Google Scholar] [CrossRef] [PubMed]
- Virant-Klun, I.; Leicht, S.; Hughes, C.; Krijgsveld, J. Identification of maturation-specific proteins by single-cell proteomics of Human oocytes. Mol. Cell. Proteomics 2016, 15, 2616–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sielaff, M.; Kuharev, J.; Bohn, T.; Hahlbrock, J.; Bopp, T.; Tenzer, S.; Distler, U. Evaluation of FASP, SP3, and iST protocols for proteomic sample preparation in the low microgram range. J. Proteome Res. 2017, 16, 4060–4072. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, K.R.; Schroll, M.M.; Hummon, A.B. Comparison of In-Solution, FASP, and S-Trap based digestion methods for bottom-up proteomic studies. J. Proteome Res. 2018, 17, 2480–2490. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhao, R.; Piehowski, P.D.; Moore, R.J.; Lim, S.; Orphan, V.J.; Pasa-Tolic, L.; Qian, W.J.; Smith, R.D.; Kelly, R.T. Subnanogram proteomics: Impact of LC column selection, MS instrumentation and data analysis strategy on proteome coverage for trace samples. Int. J. Mass Spectrom. 2018, 427, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Piehowski, P.D.; Zhao, R.; Chen, J.; Shen, Y.F.; Moore, R.J.; Shukla, A.K.; Petyuk, V.A.; Campbell-Thompson, M.; Mathews, C.E.; et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10-100 mammalian cells. Nat. Commun. 2018, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Podolak, J.; Zhao, R.; Shukla, A.K.; Moore, R.J.; Thomas, G.V.; Kelly, R.T. Proteome profiling of 1 to 5 spiked circulating tumor cells isolated from whole blood using immunodensity enrichment, laser capture microdissection, nanodroplet sample processing, and ultrasensitive nanoLC–MS. Anal. Chem. 2018, 90, 11756–11759. [Google Scholar] [CrossRef] [Green Version]
- Li, S.Y.; Plouffe, B.D.; Belov, A.M.; Ray, S.; Wang, X.Z.; Murthy, S.K.; Karger, B.L.; Ivanov, A.R. An integrated platform for isolation, processing, and mass spectrometry-based proteomic profiling of rare cells in whole blood. Mol. Cell. Proteomics 2015, 14, 1672–1683. [Google Scholar] [CrossRef] [Green Version]
- Wisniewski, J.R.; Dus, K.; Mann, M. Proteomic workflow for analysis of archival formalin-fixed and paraffin-embedded clinical samples to a depth of 10 000 proteins. Proteom. Clin. Appl. 2013, 7, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Muller, A.C.; Wagner, C.; Huber, M.L.; Rudashevskaya, E.L.; Wagner, S.N.; Bennett, K.L. Combining filter-aided sample preparation and pseudoshotgun technology to profile the proteome of a low number of early passage human melanoma cells. J. Proteome Res. 2013, 12, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Baca, M.; Desmet, G.; Ottevaere, H.; De Malsche, W. Achieving a peak capacity of 1800 using an 8 m long pillar array column. Anal. Chem. 2019, 91, 10932–10936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombard-Banek, C.; Yu, Z.; Swiercz, A.P.; Marvar, P.J.; Nemes, P. A microanalytical capillary electrophoresis mass spectrometry assay for quantifying angiotensin peptides in the brain. Anal. Bioanal. Chem. 2019, 411, 4661–4671. [Google Scholar] [CrossRef]
- Choi, S.B.; Zamarbide, M.; Manzini, M.C.; Nemes, P. Tapered-tip capillary electrophoresis nano-electrospray ionization mass spectrometry for ultrasensitive proteomics: The mouse cortex. J. Am. Soc. Mass Spectrom. 2017, 28, 597–607. [Google Scholar] [CrossRef]
- Amenson-Lamar, E.A.; Sun, L.L.; Zhang, Z.; Bohn, P.W.; Dovichi, N.J. Detection of 1 zmol injection of angiotensin using capillary zone electrophoresis coupled to a Q-Exactive HF mass spectrometer with an electrokinetically pumped sheath-flow electrospray interface. Talanta 2019, 204, 70–73. [Google Scholar] [CrossRef]
- Li, Y.H.; Champion, M.M.; Sun, L.L.; Champion, P.A.D.; Wojcik, R.; Dovichi, N.J. Capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry as an alternative proteomics platform to ultraperformance liquid chromatography-electrospray ionization-tandem mass spectrometry for samples of intermediate complexity. Anal. Chem. 2012, 84, 1617–1622. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.L.; Zhu, G.J.; Zhao, Y.M.; Yan, X.J.; Mou, S.; Dovichi, N.J. Ultrasensitive and fast bottom-up analysis of femtogram amounts of complex proteome digests. Angew. Chem.-Int. Ed. 2013, 52, 13661–13664. [Google Scholar] [CrossRef] [Green Version]
- Lombard-Banek, C.; Moody, S.A.; Nemes, P. Single-cell mass spectrometry for discovery proteomics: Quantifying translational cell heterogeneity in the 16-cell frog (Xenopus) embryo. Angew. Chem.-Int. Ed. 2016, 55, 2454–2458. [Google Scholar] [CrossRef]
- Maxwell, E.J.; Chen, D.D.Y. Twenty years of interface development for capillary electrophoresis–electrospray ionization–mass spectrometry. Anal. Chim. Acta 2008, 627, 25–33. [Google Scholar] [CrossRef]
- Lombard-Banek, C.; Reddy, S.; Moody, S.A.; Nemes, P. Label-free quantification of proteins in single embryonic cells with neural fate in the cleavage-stage frog (Xenopus laevis) embryo using capillary electrophoresis electrospray ionization high-resolution mass spectrometry (CE-ESI-HRMS). Mol. Cell. Proteomics 2016, 15, 2756–2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.Y.; Shen, X.J.; Sun, L.L. Capillary zone electrophoresis-mass spectrometry with microliter-scale loading capacity, 140 min separation window and high peak capacity for bottom-up proteomics. Analyst 2017, 142, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.J.; Spelke, D.P.; Xu, Z.C.; Kang, C.C.; Schaffer, D.V.; Herr, A.E. Single-cell western blotting. Nat. Methods 2014, 11, 749–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourelis, T.V.; Villasboas, J.C.; Jessen, E.; Dasari, S.; Dispenzieri, A.; Jevremovic, D.; Kumar, S. Mass cytometry dissects T cell heterogeneity in the immune tumor microenvironment of common dysproteinemias at diagnosis and after first line therapies. Blood Cancer J. 2019, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Marcon, E.; Jain, H.; Bhattacharya, A.; Guo, H.B.; Phanse, S.; Pu, S.Y.; Byram, G.; Collins, B.C.; Dowdell, E.; Fenner, M.; et al. Assessment of a method to characterize antibody selectivity and specificity for use in immunoprecipitation. Nat. Methods 2015, 12, 725–731. [Google Scholar] [CrossRef]
- Couvillion, S.P.; Zhu, Y.; Nagy, G.; Adkins, J.N.; Ansong, C.; Renslow, R.S.; Piehowski, P.D.; Ibrahim, Y.M.; Kelly, R.T.; Metz, T.O. New mass spectrometry technologies contributing towards comprehensive and high throughput omics analyses of single cells. Analyst 2019, 144, 794–807. [Google Scholar] [CrossRef]
- Specht, H.; Slavov, N. Transformative opportunities for single-cell proteomics. J. Proteome Res. 2018, 17, 2565–2571. [Google Scholar] [CrossRef]
- Lombard-Banek, C.; Moody, S.A.; Nemes, P. High-sensitivity mass spectrometry for probing gene translation in single embryonic cells in the early frog (Xenopus) embryo. Front. Cell Dev. Biol. 2016, 4, 100. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.L.; Dubiak, K.M.; Peuchen, E.H.; Zhang, Z.B.; Zhu, G.J.; Huber, P.W.; Dovichi, N.J. Single cell proteomics using frog (Xenopus laevis) blastomeres isolated from early stage embryos, which form a geometric progression in protein content. Anal. Chem. 2016, 88, 6653–6657. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Zhang, Z.; Liu, Y.Z.; Gao, Y.; Gu, J.K. Recent advances in single-cell analysis by mass spectrometry. Analyst 2019, 144, 824–845. [Google Scholar] [CrossRef]
- Lombard-Banek, C.; Moody, S.A.; Manzin, M.C.; Nemes, P. Microsampling capillary electrophoresis mass spectrometry enables single-cell proteomics in complex tissues: Developing cell clones in live Xenopus laevis and Zebrafish embryos. Anal. Chem. 2019, 91, 4797–4805. [Google Scholar] [CrossRef] [PubMed]
- Lombard-Banek, C.; Choi, S.B.; Nemes, P. Single-cell proteomics in complex tissues using microprobe capillary electrophoresis mass spectrometry. Methods Enzymol. 2019, 628, 263–292. [Google Scholar] [PubMed]
- Li, Z.Y.; Huang, M.; Wang, X.K.; Zhu, Y.; Li, J.S.; Wong, C.C.L.; Fang, Q. Nanoliter-scale oil-air-droplet chip-based single cell proteomic analysis. Anal. Chem. 2018, 90, 5430–5438. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Clair, G.; Chrisler, W.B.; Shen, Y.F.; Zhao, R.; Shukla, A.K.; Moore, R.J.; Misra, R.S.; Pryhuber, G.S.; Smith, R.D.; et al. Proteomic analysis of single mammalian cells enabled by microfluidic nanodroplet sample preparation and ultrasensitive NanoLC-MS. Angew. Chem. Int. Ed. 2018, 57, 12370–12374. [Google Scholar] [CrossRef] [PubMed]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Zilionis, R.; Nainys, J.; Veres, A.; Savova, V.; Zemmour, D.; Klein, A.M.; Mazutis, L. Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc. 2017, 12, 44–73. [Google Scholar] [CrossRef]
- Ting, L.; Rad, R.; Gygi, S.P.; Haas, W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 2011, 8, 937–940. [Google Scholar] [CrossRef] [Green Version]
- McAlister, G.C.; Nusinow, D.P.; Jedrychowski, M.P.; Wuhr, M.; Huttlin, E.L.; Erickson, B.K.; Rad, R.; Haas, W.; Gygi, S.P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014, 86, 7150–7158. [Google Scholar] [CrossRef]
- Budnik, B.; Levy, E.; Harmange, G.; Slavov, N. SCoPE-MS: Mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19, 161. [Google Scholar] [CrossRef] [Green Version]
- Dou, M.W.; Clair, G.; Tsai, C.F.; Xu, K.R.; Chrisler, W.B.; Sontag, R.L.; Zhao, R.; Moore, R.J.; Liu, T.; Pasa-Tolic, L.; et al. High-throughput single cell proteomics enabled by multiplex isobaric labeling in a nanodroplet sample preparation platform. Anal. Chem. 2019, 91, 13119–13127. [Google Scholar] [CrossRef]
- Gong, H.B.; Do, D.; Ramakrishnan, R. Single-cell mRNA-seq using the Fluidigm C1 system and integrated fluidics circuits. In Gene Expression Analysis: Methods and Protocols; Raghavachari, N., GarciaReyero, N., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2018; Volume 1783, pp. 193–207. [Google Scholar]
- McAlister, G.C.; Huttlin, E.L.; Haas, W.; Ting, L.; Jedrychowski, M.P.; Rogers, J.C.; Kuhn, K.; Pike, I.; Grothe, R.A.; Blethrow, J.D.; et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem. 2012, 84, 7469–7478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, C.R.; Bird, G.H.; Wuhr, M.; Erickson, B.K.; Rad, R.; Walensky, L.D.; Gygi, S.P.; Haas, W. Generation of multiple reporter ions from a single isobaric reagent increases multiplexing capacity for quantitative proteomics. Anal. Chem. 2015, 87, 9855–9863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Q.; Bettini, E.; Paczkowski, P.; Ng, C.; Kaiser, A.; McConnell, T.; Kodrasi, O.; Quigley, M.F.; Heath, J.; Fan, R.; et al. Single-cell multiplexed cytokine profiling of CD19 CAR-T cells reveals a diverse landscape of polyfunctional antigen-specific response. J. Immunother. Cancer 2017, 5, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.S.; Abernathy, M.; Richardson, D.D.; Rouse, J.C.; Sperry, J.B.; Swann, P.; Wypych, J.; Yu, C.; Zang, L.; Deshpande, R. A View on the importance of “multi-attribute method” for measuring purity of biopharmaceuticals and improving overall control strategy. AAPS J. 2018, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogstad, S.; Yan, H.H.; Wang, X.S.; Powers, D.; Brorson, K.; Damdinsuren, B.; Lee, S. Multi-attribute method for quality control of therapeutic proteins. Anal. Chem. 2019, 91, 14170–14177. [Google Scholar] [CrossRef]
- Bereman, M.S.; Johnson, R.; Bollinger, J.; Boss, Y.; Shulman, N.; MacLean, B.; Hoofnagle, A.N.; MacCoss, M.J. Implementation of statistical process control for proteomic experiments via LC MS/MS. J. Am. Soc. Mass Spectrom. 2014, 25, 581–587. [Google Scholar] [CrossRef] [Green Version]
- Bereman, M.S. Tools for monitoring system suitability in LC MS/MS centric proteomic experiments. Proteomics 2015, 15, 891–902. [Google Scholar] [CrossRef]
- Bittremieux, W.; Walzer, M.; Tenzer, S.; Zhu, W.M.; Salek, R.M.; Eisenacher, M.; Tabb, D.L. The Human proteome organization-proteomics standards initiative quality control working group: Making quality control more accessible for biological mass spectrometry. Anal. Chem. 2017, 89, 4474–4479. [Google Scholar] [CrossRef]
- Ivanov, A.R.; Colangelo, C.M.; Dufresne, C.P.; Friedman, D.B.; Lilley, K.S.; Mechtler, K.; Phinney, B.S.; Rose, K.L.; Rudnick, P.A.; Searle, B.C.; et al. Interlaboratory studies and initiatives developing standards for proteomics. Proteomics 2013, 13, 904–909. [Google Scholar] [CrossRef] [Green Version]
- Bittremieux, W.; Tabb, D.L.; Impens, F.; Staes, A.; Timmerman, E.; Martens, L.; Laukens, K. Quality control in mass spectrometry-based proteomics. Mass Spectrom. Rev. 2018, 37, 697–711. [Google Scholar] [CrossRef]
- Rudnick, P.A.; Clauser, K.R.; Kilpatrick, L.E.; Tchekhovskoi, D.V.; Neta, P.; Blonder, N.; Billheimer, D.D.; Blackman, R.K.; Bunk, D.M.; Cardasis, H.L.; et al. Performance metrics for liquid chromatography-tandem mass spectrometry systems in proteomics analyses. Mol. Cell. Proteomics 2010, 9, 225–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocher, T.; Pichler, P.; Swart, R.; Mechtler, K. Quality control in LC-MS/MS. Proteomics 2011, 11, 1026–1030. [Google Scholar] [CrossRef]
- Klimek, J.; Eddes, J.S.; Hohmann, L.; Jackson, J.; Peterson, A.; Letarte, S.; Gafken, P.R.; Katz, J.E.; Mallick, P.; Lee, H.; et al. The standard protein mix database: A diverse data set to assist in the production of improved peptide and protein identification software tools. J. Proteome Res. 2008, 7, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulovich, A.G.; Billheimer, D.; Ham, A.J.L.; Vega-Montoto, L.; Rudnick, P.A.; Tabb, D.L.; Wang, P.; Blackman, R.K.; Bunk, D.M.; Cardasis, H.L.; et al. Interlaboratory study characterizing a yeast performance standard for benchmarking LC-MS platform performance. Mol. Cell. Proteomics 2010, 9, 242–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beasley-Green, A.; Bunk, D.; Rudnick, P.; Kilpatrick, L.; Phinney, K. A proteomics performance standard to support measurement quality in proteomics. Proteomics 2012, 12, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Beri, J.; Rosenblatt, M.M.; Strauss, E.; Urh, M.; Bereman, M.S. Reagent for evaluating liquid chromatography–tandem mass spectrometry (LC-MS/MS) performance in bottom-up proteomic experiments. Anal. Chem. 2015, 87, 11635–11640. [Google Scholar] [CrossRef]
- Wong, C.C.L.; Cociorva, D.; Miller, C.A.; Schmidt, A.; Monell, C.; Aebersold, K.; Yates, J.R. Proteomics of Pyrococcus furiosus (Pfu): Identification of extracted proteins by three independent methods. J. Proteome Res. 2013, 12, 763–770. [Google Scholar] [CrossRef] [Green Version]
- Scheltema, R.A.; Mann, M. SprayQc: A real-time LC–MS/MS quality monitoring system to maximize uptime using off the shelf components. J. Proteome Res. 2012, 11, 3458–3466. [Google Scholar] [CrossRef]
- Nemes, P.; Marginean, I.; Vertes, A. Spraying mode effect on droplet formation and ion chemistry in electrosprays. Anal. Chem. 2007, 79, 3105–3116. [Google Scholar] [CrossRef]
- Broudy, D.; Killeen, T.; Choi, M.; Shulman, N.; Mani, D.R.; Abbatiello, S.E.; Mani, D.; Ahmad, R.; Sahu, A.K.; Schilling, B.; et al. A framework for installable external tools in Skyline. Bioinformatics 2014, 30, 2521–2523. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.Q.; Polzin, K.O.; Dasari, S.; Charnbers, M.C.; Schilling, B.; Gibson, B.W.; Tran, B.Q.; Vega–Montoto, L.; Liebler, D.C.; Tabb, D.L. QuaMeter: Multivendor performance metrics for LC-MS/MS proteomics instrumentation. Anal. Chem. 2012, 84, 5845–5850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogu, E.; Mohammad-Taheri, S.; Abbatiello, S.E.; Bereman, M.S.; Maclean, B.; Schilling, B.; Vitek, O. MSstatsQC: Longitudinal system suitability monitoring and quality control for targeted proteomic experiments. Mol. Cell. Proteomics 2017, 16, 1335–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielow, C.; Mastrobuoni, G.; Kempa, S. Proteomics quality control: Quality control software for MaxQuant results. J. Proteome Res. 2015, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Paulo, J.A.; O’Connell, J.D.; Gygi, S.P. A triple Kkockout (TKO) proteomics standard for diagnosing ion interference in isobaric labeling experiments. J. Am. Soc. Mass Spectrom. 2016, 27, 1620–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulo, J.A.; Navarrete-Perea, J.; Thakurta, S.G.; Gygi, S.P. TKO6: A peptide standard to assess interference for unit-resolved isobaric labeling platforms. J. Proteome Res. 2019, 18, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Gygi, J.P.; Yu, Q.; Navarrete-Perea, J.; Rad, R.; Gygi, S.P.; Paulo, J.A. Web-based search tool for visualizing instrument performance using the triple knockout (TKO) proteome standard. J. Proteome Res. 2019, 18, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Schiel, J.E.; Mires, L.A.; Davis, D.L. State-of-the-art and emerging technologies for therapeutic monoclonal antibody characterization, Vol 1–Monoclonal antibody therapeutics: Structure, function, and regulatory space. In State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization, Vol 1–Monoclonal Antibody Therapeutics: Structure, Function, and Regulatory Space; Schiel, J.E., Davis, D.L., Borisov, O.V., Eds.; Amer Chemical Soc: Washington, DC, USA, 2014; Volume 1176, pp. 1–165. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Preparation | Digestion Location | Lysis on Device | Reagent Compatibility | Sample Input | Online Clean-up | Refs. |
|---|---|---|---|---|---|---|
| FASP | On filter | No | Wide variety of detergents and reagents | >20 μg | No | [73] |
| S-Trap | On filter | No | Wide variety of detergents and reagents | ~500 ng–500 μg | Yes | [78] |
| iST | On filter | Yes | Cleavable detergent only | <1 μg–500 μg | Yes | [77] |
| Streamlined iST | On filter | Yes | Cleavable detergent only | <2 μg | Yes | [80] |
| SP3 | In tube | Yes | Wide variety of detergents and reagents | 100 ng–500 μg | No | [76,81] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lombard-Banek, C.; Schiel, J.E. Mass Spectrometry Advances and Perspectives for the Characterization of Emerging Adoptive Cell Therapies. Molecules 2020, 25, 1396. https://doi.org/10.3390/molecules25061396
Lombard-Banek C, Schiel JE. Mass Spectrometry Advances and Perspectives for the Characterization of Emerging Adoptive Cell Therapies. Molecules. 2020; 25(6):1396. https://doi.org/10.3390/molecules25061396
Chicago/Turabian StyleLombard-Banek, Camille, and John E. Schiel. 2020. "Mass Spectrometry Advances and Perspectives for the Characterization of Emerging Adoptive Cell Therapies" Molecules 25, no. 6: 1396. https://doi.org/10.3390/molecules25061396
APA StyleLombard-Banek, C., & Schiel, J. E. (2020). Mass Spectrometry Advances and Perspectives for the Characterization of Emerging Adoptive Cell Therapies. Molecules, 25(6), 1396. https://doi.org/10.3390/molecules25061396