Profiling of Seed Proteome in Pea (Pisum sativum L.) Lines Characterized with High and Low Responsivity to Combined Inoculation with Nodule Bacteria and Arbuscular Mycorrhizal Fungi

 , , ,
, , ,  and
and
Abstract
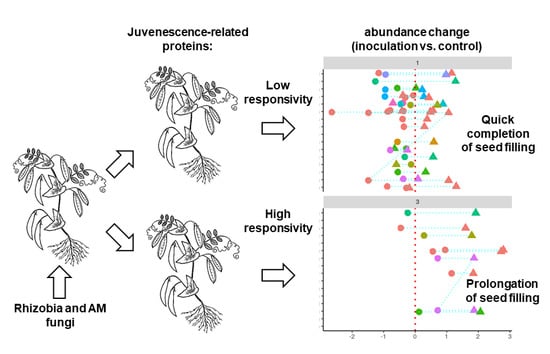
:
1. Introduction
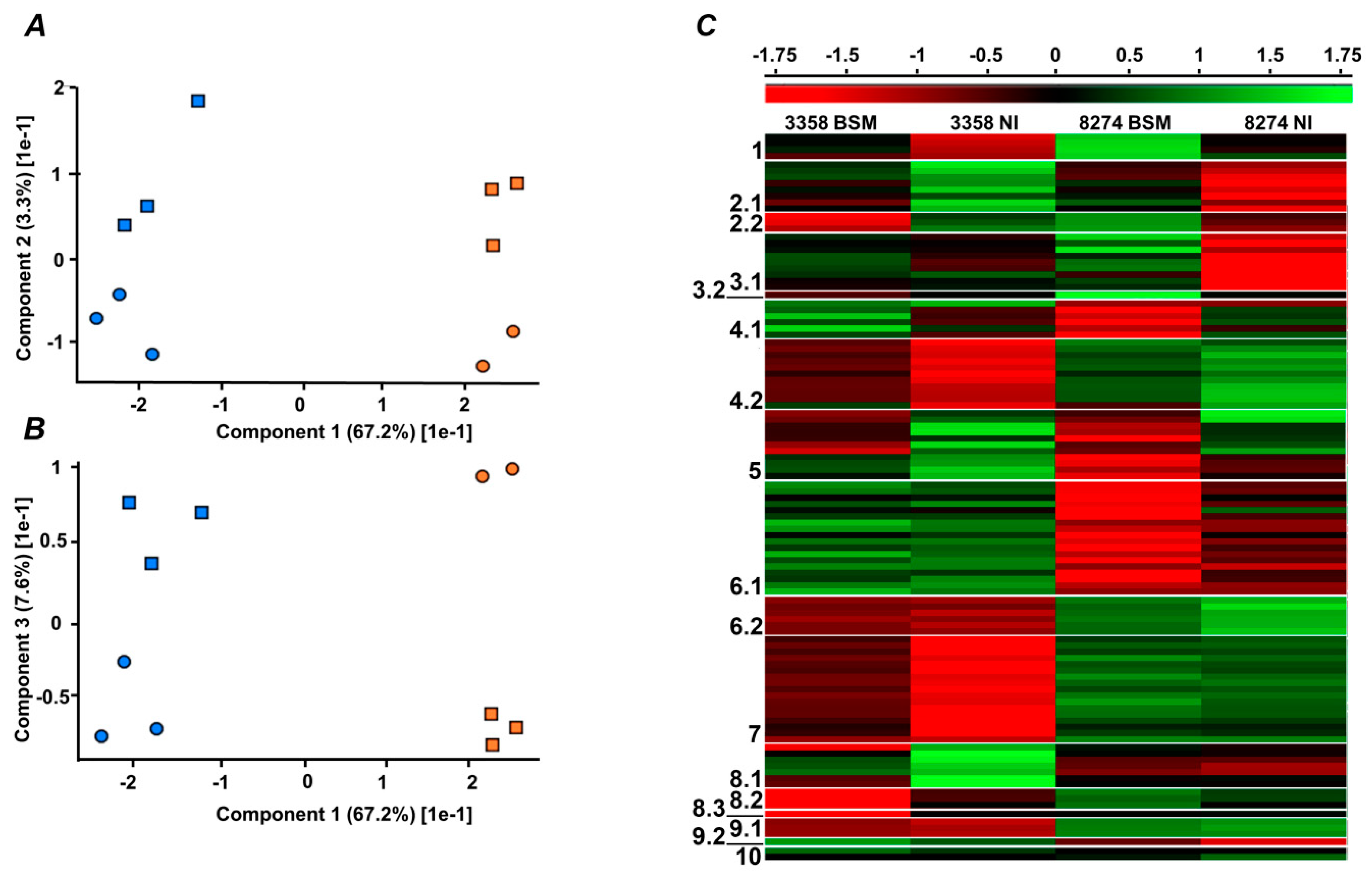
2. Results
2.1. Biomass Gain and Seed Productivity
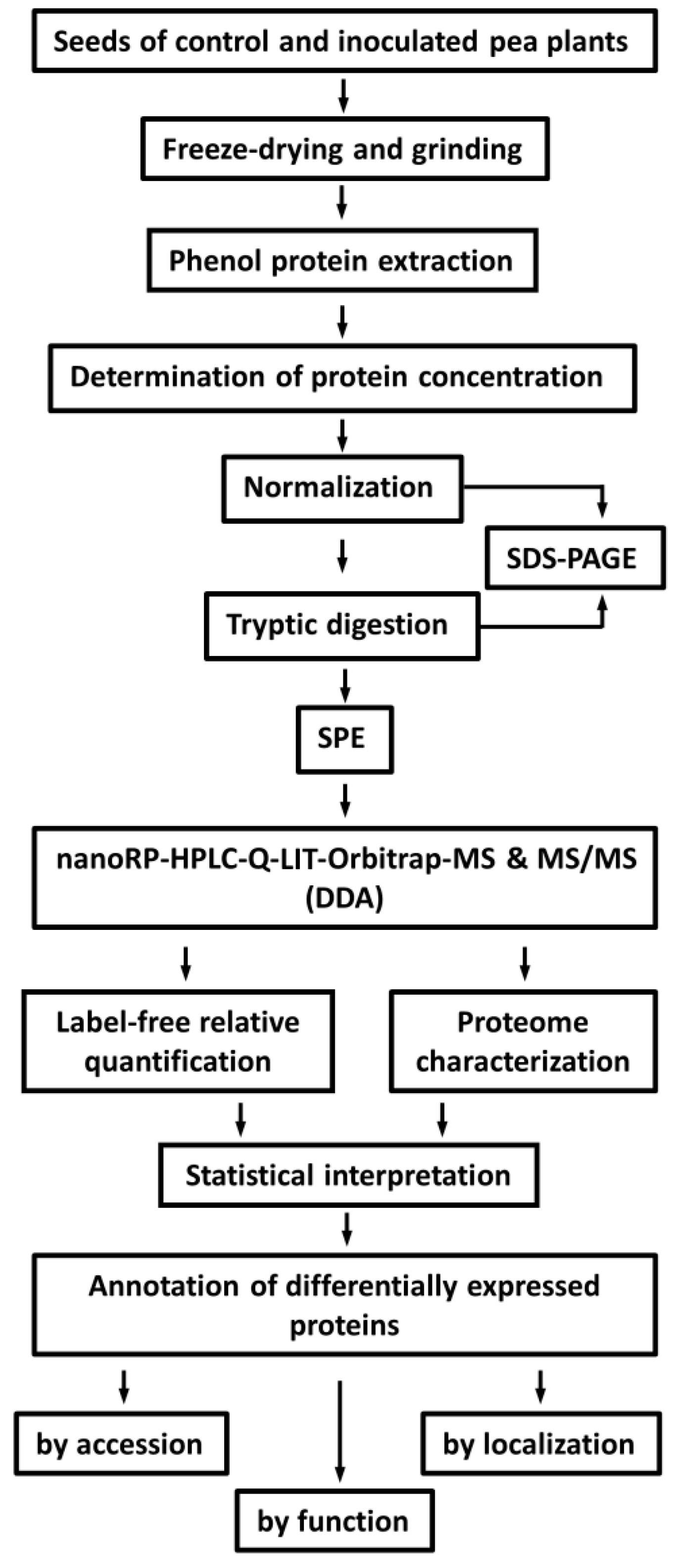
2.2. Protein Isolation and Tryptic Digestion
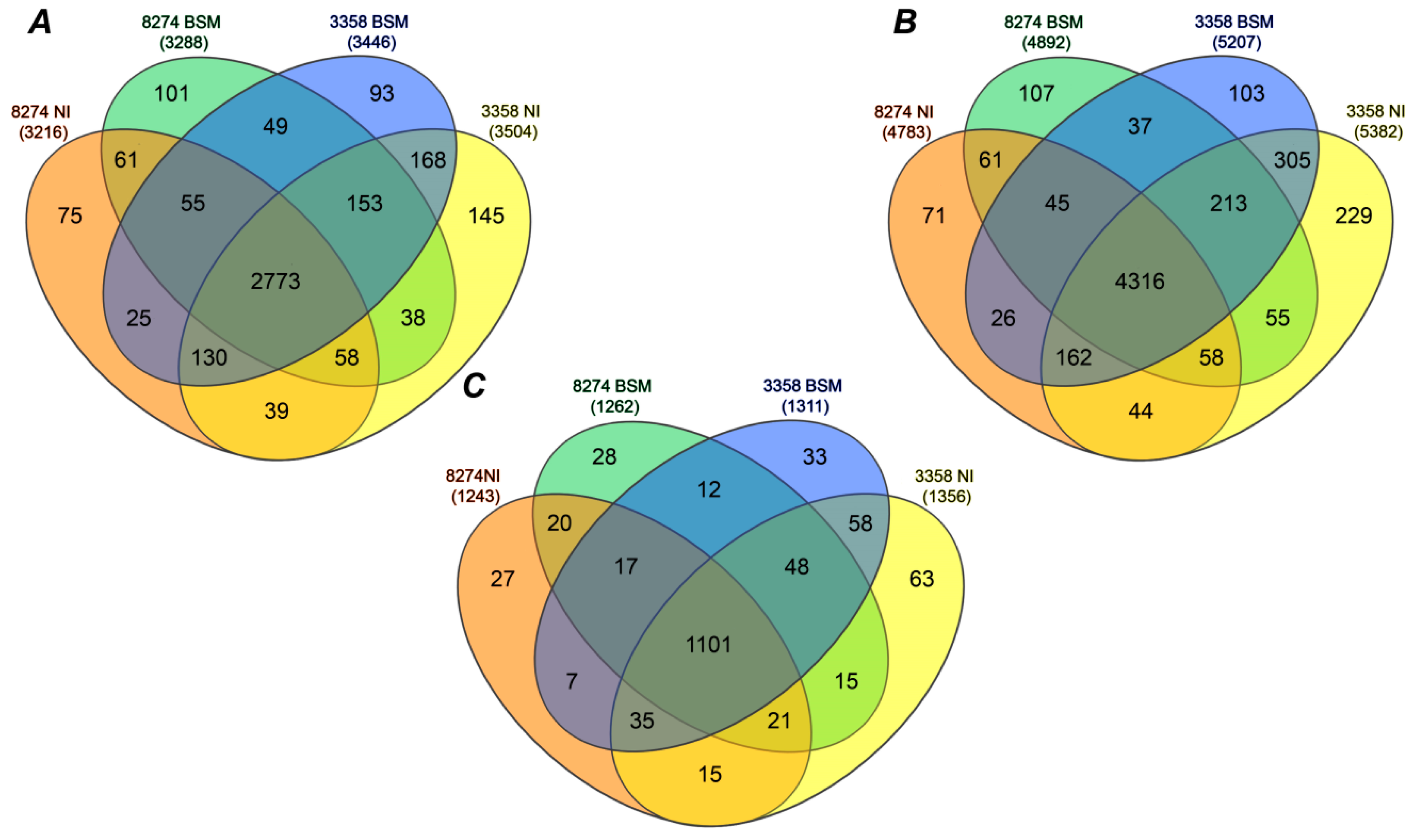
2.3. Identification of Seed Proteins
2.4. Label-Free Quantification
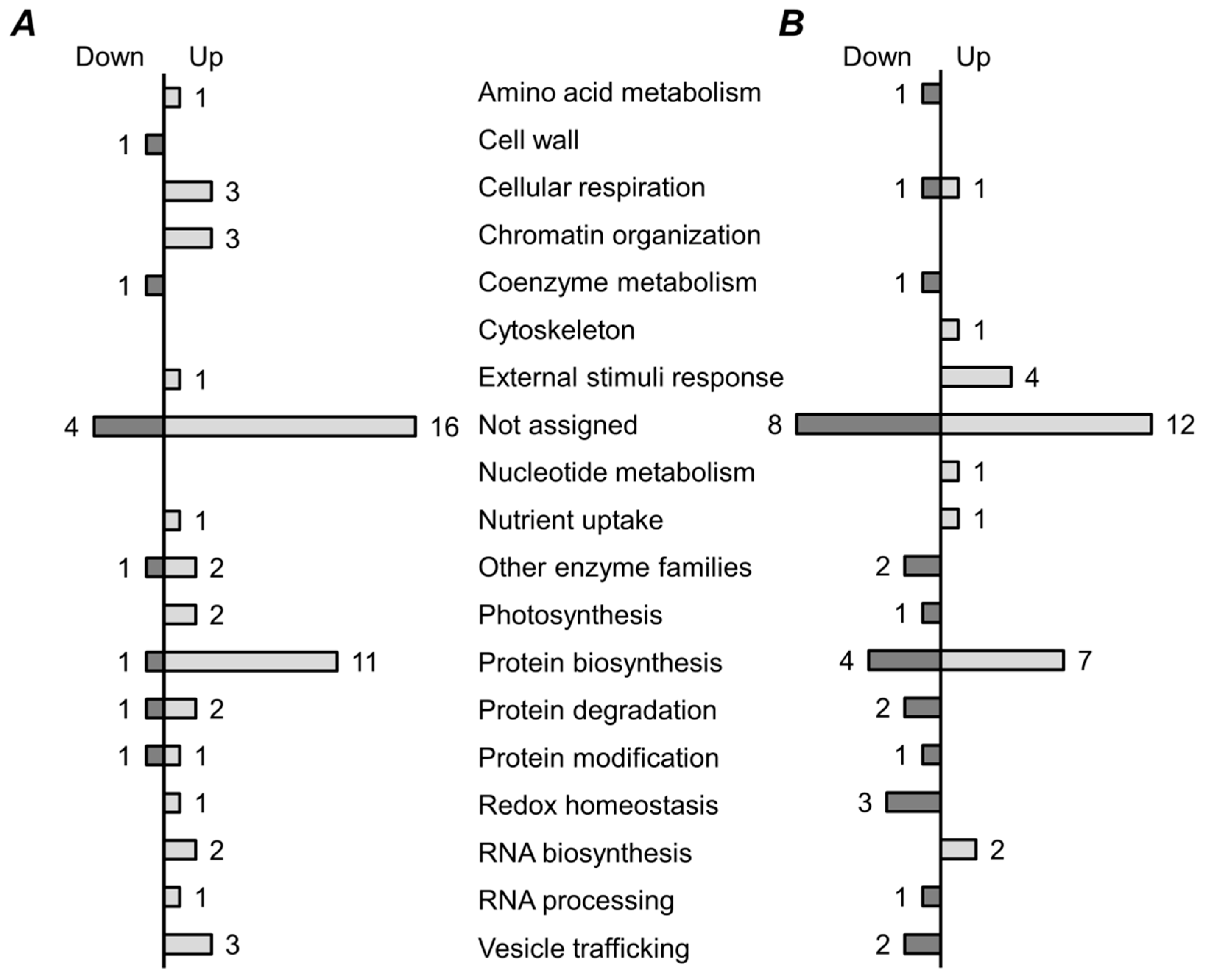
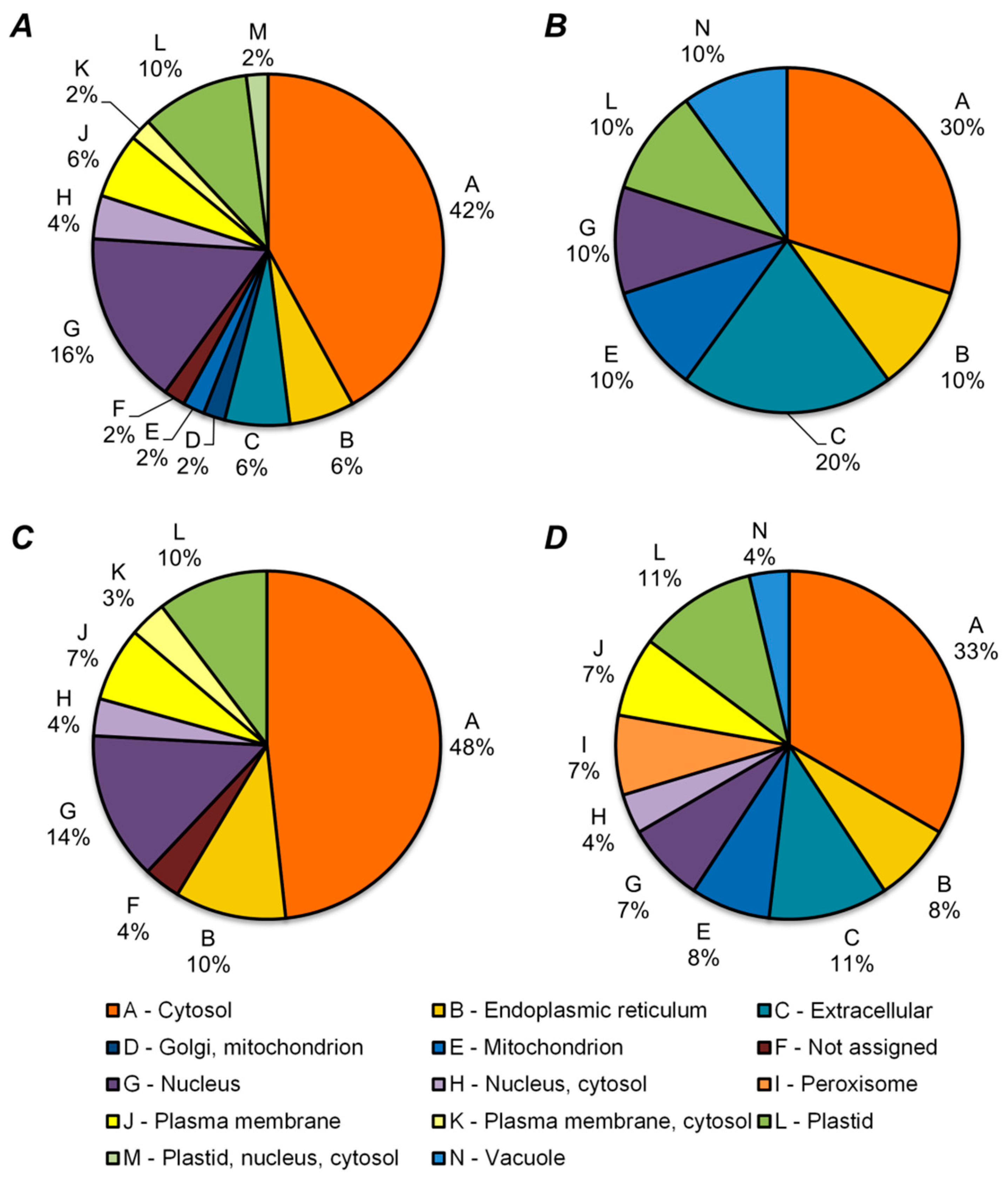
2.5. Functional Annotation of Differentially Regulated Proteins
3. Discussion
3.1. Complex Inoculation Affects Seed Productivity only in the High-EIBSM Line K-8274
3.2. Differences in Protein Expression Patterns between the Pea Lines with High and Low EIBSM
3.3. Response of High- and Low-EIBSM Pea Lines to Inoculation with Rhizobia and Arbuscular Mycorrhiza
3.4. Ecological and Agricultural Aspects of the High-EIBSM Trait
4. Materials and Methods
4.1. Reagents
4.2. AM Fungal Inoculum
4.3. Plant Experiments and Characterization of Biomass Gain and Seed Productivity
4.4. Protein Isolation
4.5. Tryptic Digestion
4.6. LC-MS Experiments
4.7. Data Analysis and Post-Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shtark, O.Y.; Borisov, A.Y.; Zhukov, V.A.; Tikhonovich, I.A. Mutually beneficial legume symbioses with soil microbes and their potential for plant production. Symbiosis 2012, 58, 51–62. [Google Scholar] [CrossRef]
- Oldroyd, G.E.D. Speak, friend, and enter: Signalling systems that promote beneficial symbiotic associations in plants. Nat. Rev. Microbiol. 2013, 11, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.P. Carbon and Nitrogen Metabolism in Legume Nodules. In Nitrogen-Fixing Leguminous Symbioses; Springer: Dordrecht, The Netherlands, 2008; pp. 293–320. [Google Scholar]
- Smith, S.E.; Andrew Smith, F. Roles of Arbuscular Mycorrhizas in Plant Nutrition and Growth: New Paradigms from Cellular to Ecosystem Scales. Annu. Rev. Plant Biol. 2011, 62, 227–250. [Google Scholar] [CrossRef] [PubMed]
- Velázquez, E.; García-Fraile, P.; Ramírez-Bahena, M.-H.; Rivas, R.; Martínez-Molina, E. Current Status of the Taxonomy of Bacteria Able to Establish Nitrogen-Fixing Legume Symbiosis. In Microbes for Legume Improvement; Springer International Publishing: Cham, Germany, 2017; pp. 1–43. [Google Scholar]
- Glick, B.R. Plant Growth-Promoting Bacteria: Mechanisms and Applications. Scientifica 2012, 2012, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pii, Y.; Mimmo, T.; Tomasi, N.; Terzano, R.; Cesco, S.; Crecchio, C. Microbial interactions in the rhizosphere: Beneficial influences of plant growth-promoting rhizobacteria on nutrient acquisition process. A review. Biol. Fertil. Soils 2015, 51, 403–415. [Google Scholar] [CrossRef]
- Lugtenberg, B.; Rozen, D.E.; Kamilova, F. Wars between microbes on roots and fruits. F1000Research 2017, 6, 343. [Google Scholar] [CrossRef] [PubMed]
- Muleta, D. Legume Response to Arbuscular Mycorrhizal Fungi Inoculation in Sustainable Agriculture. In Microbes for Legume Improvement; Springer International Publishing: Cham, Germany, 2017; pp. 227–260. [Google Scholar]
- Barea, J.-M.; Azcón, R.; Azcón-Aguilar, C. Mycorrhizosphere interactions to improve plant fitness and soil quality. Antonie Van Leeuwenhoek 2002, 81, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Zaidi, A.; Musarrat, J. Microbes for Legume Improvement; Springer-Verlag: Wien, Austria, 2017; ISBN 978-3-319-59173-5. [Google Scholar]
- Shtark, O.Y.; Zhukov, V.; Sulima, A.S.; Singh, R.; Naumkina, T.S.; Akhtemova, G.A.; Borisov, A.Y. Prospects for the use of multi-component symbiotic systems of the Legumes. Ecol. Genet. 2015, 13, 33–46. [Google Scholar] [CrossRef]
- Shtark, O.Y.; Borisov, A.Y.; Zhukov, V.A.; Provorov, N.A.; Tikhonovich, I.A. Intimate associations of beneficial soil microbes with the host plants. In Soil Microbiology and Sustainable Crop Production; Springer: Dordrecht, The Netherlands, 2010; pp. 119–196. ISBN 978-90-481-9478-0. [Google Scholar]
- Van der Heijden, M.G.A.; de Bruin, S.; Luckerhoff, L.; van Logtestijn, R.S.P.; Schlaeppi, K. A widespread plant-fungal-bacterial symbiosis promotes plant biodiversity, plant nutrition and seedling recruitment. ISME J. 2016, 10, 389–399. [Google Scholar] [CrossRef]
- Lüscher, A.; Mueller-Harvey, I.; Soussana, J.F.; Rees, R.M.; Peyraud, J.L. Potential of Legume-Based Grassland-Livestock Systems in Europe: A Review. Grass Forage Sci. 2014, 69, 206–228. [Google Scholar] [CrossRef] [PubMed]
- Oldroyd, G.E.; Dixon, R. Biotechnological solutions to the nitrogen problem. Curr. Opin. Biotechnol. 2014, 26, 19–24. [Google Scholar] [CrossRef]
- Limpens, E.; Moling, S.; Hooiveld, G.; Pereira, P.A.; Bisseling, T.; Becker, J.D.; Küster, H. Cell- and Tissue-Specific Transcriptome Analyses of Medicago truncatula Root Nodules. PLoS ONE 2013, 8, e64377. [Google Scholar] [CrossRef] [PubMed]
- Camps, C.; Jardinaud, M.; Rengel, D.; Carrère, S.; Hervé, C.; Debellé, F.; Gamas, P.; Bensmihen, S.; Gough, C. Combined genetic and transcriptomic analysis reveals three major signalling pathways activated by Myc-LCOs in Medicago truncatula. New Phytol. 2015, 208, 224–240. [Google Scholar] [CrossRef] [PubMed]
- Küster, H.; Vieweg, M.F.; Manthey, K.; Baier, M.C.; Hohnjec, N.; Perlick, A.M. Identification and expression regulation of symbiotically activated legume genes. Phytochemistry 2007, 68, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Afkhami, M.E.; Stinchcombe, J.R. Multiple mutualist effects on genomewide expression in the tripartite association between Medicago truncatula, nitrogen-fixing bacteria and mycorrhizal fungi. Mol. Ecol. 2016, 25, 4946–4962. [Google Scholar] [CrossRef]
- Nielsen, J.; Oliver, S. The next wave in metabolome analysis. Trends Biotechnol. 2005, 23, 544–546. [Google Scholar] [CrossRef]
- Duranti, M. Grain Legume Proteins and Nutraceutical Properties. Fitoterapia 2006, 77, 67–82. [Google Scholar] [CrossRef]
- Gillet, L.C.; Leitner, A.; Aebersold, R. Mass Spectrometry Applied to Bottom-Up Proteomics: Entering the High-Throughput Era for Hypothesis Testing. Annu. Rev. Anal. Chem. 2016, 9, 449–472. [Google Scholar] [CrossRef] [PubMed]
- Paudel, G.; Bilova, T.; Schmidt, R.; Greifenhagen, U.; Berger, R.; Tarakhovskaya, E.; Stöckhardt, S.; Balcke, G.U.; Humbeck, K.; Brandt, W.; et al. Osmotic stress is accompanied by protein glycation in Arabidopsis thaliana. J. Exp. Bot. 2016, erw395. [Google Scholar] [CrossRef]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bügel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced glycation endproducts in food and their effects on health. Food Chem. Toxicol. 2013, 60, 10–37. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, L.M.; Kukalev, A.S.; Ushakov, K.V.; Tsyganov, V.E.; Provorov, N.A.; Borisov, A.Y.; Tikhonovich, I. Genetic variability of garden pea (Pisum sativum, L.) for symbiotic capacities. Pisum. Genet. 1999, 31, 44–45. [Google Scholar]
- Borisov, A.Y.; Tsyganov, V.E.; Shtark, O.Y.; Jacobi, L.M.; Naumkina, T.S.; Serdyuk, V.P.; Vishnyakova, M.A. Pea: Symbiotic effectiveness. Cat. World Collect. VIR 2002, 728, 1–29. [Google Scholar]
- Chebotar, V.K.; Kazakov, A.E.; Erofeev, S.V.; Danilova, T.N.; Naumkina, T.S.; Shtark, O.Y.; Tikhonovich, I.A.; Borisov, A.Y. Method of production of complex microbial fertilizer. RF Patent No. 2318784, 30 Matrch 2006. [Google Scholar]
- Shtark, O.Y.; Danilova, T.N.; Naumkina, T.S.; Vasilchikov, A.G.; Chebotar, V.K.; Kazakov, A.E.; Zhernakov, A.I.; Nemankin, T.A.; Prilepskaya, N.A.; Borisov, A.U.; et al. Analysis Of Pea (Pisum Sativum L.) Source Material For Breeding Of Cultivars With High Symbiotic Potential And Choice Of Criteria For Its Evaluation. Ecol. Genet. 2006, 4, 22–28. [Google Scholar] [CrossRef]
- Borisov, A.Y.; Naumkina, T.S.; Shtark, O.Y.; Danilova, T.N.; Tsyganov, V.E. Effectiveness of combined inoculation of pea (Pisum sativum L.) with arbuscular mycorrhizal fungu and rhizobia. Proc. Russ. Acad. Agric. Sci. 2004, 2, 12–14. [Google Scholar]
- Mamontova, T.; Lukasheva, E.; Mavropolo-Stolyarenko, G.; Proksch, C.; Bilova, T.; Kim, A.; Babakov, V.; Grishina, T.; Hoehenwarter, W.; Medvedev, S.; et al. Proteome Map of Pea (Pisum sativum L.) Embryos Containing Different Amounts of Residual Chlorophylls. Int. J. Mol. Sci. 2018, 19, 4066. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Blüher, M.; Hoffmann, R. Glycation sites of human plasma proteins are affected to different extents by hyperglycemic conditions in type 2 diabetes mellitus. Anal. Bioanal. Chem. 2014, 406, 5755–5763. [Google Scholar] [CrossRef] [PubMed]
- Barac, M.; Cabrilo, S.; Pesic, M.; Stanojevic, S.; Zilic, S.; Macej, O.; Ristic, N.; Barac, M.; Cabrilo, S.; Pesic, M.; et al. Profile and Functional Properties of Seed Proteins from Six Pea (Pisum sativum) Genotypes. Int. J. Mol. Sci. 2010, 11, 4973–4990. [Google Scholar] [CrossRef]
- Matamoros, M.A.; Kim, A.; Peñuelas, M.; Ihling, C.; Griesser, E.; Hoffmann, R.; Fedorova, M.; Frolov, A.; Becana, M. Protein Carbonylation and Glycation in Legume Nodules. Plant Physiol. 2018, 177, 1510–1528. [Google Scholar] [CrossRef]
- Lohse, M.; Nagel, A.; Herter, T.; May, P.; Schroda, M.; Zrenner, R.; Tohge, T.; Fernie, A.R.; Stitt, M.; Usadel, B. Mercator: A fast and simple web server for genome scale functional annotation of plant sequence data. Plant Cell Environ. 2014, 37, 1250–1258. [Google Scholar] [CrossRef]
- Chen, M.; Thelen, J.J. The Plastid Isoform of Triose Phosphate Isomerase Is Required for the Postgerminative Transition from Heterotrophic to Autotrophic Growth in Arabidopsis. Plant Cell 2010, 22, 77–90. [Google Scholar] [CrossRef]
- Dure, L.; Greenway, S.C.; Galau, G.A. Developmental biochemistry of cottonseed embryogenesis and germination: Changing messenger ribonucleic acid populations as shown by in vitro and in vivo protein synthesis. Biochemistry 1981, 20, 4162–4168. [Google Scholar] [CrossRef] [PubMed]
- Min, C.W.; Lee, H.S.; Cheon, Y.E.; Han, W.Y.; Ko, J.M.; Kang, H.W.; Kim, Y.C.; Agrawal, G.K.; Rakwal, R.; Gupta, R.; et al. In-depth proteomic analysis of Glycine max seeds during controlled deterioration treatment reveals a shift in seed metabolism. J. Proteomics 2017, 169, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Ranjbar Sistani, N.; Kaul, H.-P.; Desalegn, G.; Wienkoop, S. Rhizobium Impacts on Seed Productivity, Quality, and Protection of Pisum sativum upon Disease Stress Caused by Didymella pinodes: Phenotypic, Proteomic, and Metabolomic Traits. Front. Plant Sci. 2017, 8, 1961. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Mamontova, T.; Ihling, C.; Lukasheva, E.; Bankin, M.; Chantseva, V.; Vikhnina, M.; Soboleva, A.; Shumilina, J.; Osmolovskaya, N.; et al. Mining seed proteome: From protein dynamics to modification profiles. Biol. Commun. 2018, 63, 43–58. [Google Scholar] [CrossRef]
- Borisov, A.Y.; Danilova, T.N.; Koroleva, T.A.; Naumkina, T.S.; Pavlova, Z.B.; Pinaev, A.G.; Shtark, O.Y.; Tsyganov, V.E.; Voroshilova, V.A.; Zhernakov, A.I.; et al. Pea (Pisum sativum L.) regulatory genes controlling development of nitrogen-fixing nodule and arbuscular mycorrhiza: Fundamentals and application. Biologia 2004, 59, 137–144. [Google Scholar]
- Desalegn, G.; Turetschek, R.; Kaul, H.-P.; Wienkoop, S. Microbial symbionts affect Pisum sativum proteome and metabolome under Didymella pinodes infection. J. Proteomics 2016, 143, 173–187. [Google Scholar] [CrossRef]
- Hooper, C.M.; Tanz, S.K.; Castleden, I.R.; Vacher, M.A.; Small, I.D.; Millar, A.H. SUBAcon: A consensus algorithm for unifying the subcellular localization data of the Arabidopsis proteome. Bioinformatics 2014, 30, 3356–3364. [Google Scholar] [CrossRef] [PubMed]
- Turetschek, R.; Lyon, D.; Desalegn, G.; Kaul, H.-P.; Wienkoop, S. A Proteomic Workflow Using High-Throughput De Novo Sequencing Towards Complementation of Genome Information for Improved Comparative Crop Science; In Humana Press: New York, NY, USA, 2016; pp. 233–243. [Google Scholar]
- Ellis, T.H.N.; Hofer, J.M.I.; Timmerman-Vaughan, G.M.; Coyne, C.J.; Hellens, R.P. Mendel, 150 years on. Trends Plant Sci. 2011, 16, 590–596. [Google Scholar] [CrossRef]
- Smolikova, G.; Dolgikh, E.; Vikhnina, M.; Frolov, A.; Medvedev, S. Genetic and Hormonal Regulation of Chlorophyll Degradation during Maturation of Seeds with Green Embryos. Int. J. Mol. Sci. 2017, 18, 1993. [Google Scholar] [CrossRef]
- Almoguera, C.; Jordano, J. Developmental and environmental concurrent expression of sunflower dry-seed-stored low-molecular-weight heat-shock protein and Lea mRNAs. Plant Mol. Biol. 1992, 19, 781–792. [Google Scholar] [CrossRef]
- Kalemba, E.M.; Pukacka, S. Changes in late embryogenesis abundant proteins and a small heat shock protein during storage of beech (Fagus sylvatica L.) seeds. Environ. Exp. Bot. 2008, 63, 274–280. [Google Scholar] [CrossRef]
- Pawłowski, T.A. Proteome analysis of Norway maple (Acer platanoides L.) seeds dormancy breaking and germination: Influence of abscisic and gibberellic acids. BMC Plant Biol. 2009, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Fung, P.; Nishimura, N.; Jensen, D.R.; Fujii, H.; Zhao, Y.; Lumba, S.; Santiago, J.; Rodrigues, A.; Chow, T.-F.F.; et al. Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science 2009, 324, 1068–1071. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, M.; Jacquin, F.; Cassecuelle, F.; Savois, V.; Belghazi, M.; Aubert, G.; Quillien, L.; Huart, M.; Marget, P.; Burstin, J. A PQL (protein quantity loci) analysis of mature pea seed proteins identifies loci determining seed protein composition. Proteomics 2011, 11, 1581–1594. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, T.; Tanaka, K.; Monma, M.; Kawamura, Y.; Saio, K. Phosphoenolpyruvate Carboxylase Level in Soybean Seed Highly Correlates to Its Contents of Protein and Lipid. Agric. Biol. Chem. 1989, 53, 885–887. [Google Scholar]
- Benedito, V.A.; Torres-Jerez, I.; Murray, J.D.; Andriankaja, A.; Allen, S.; Kakar, K.; Wandrey, M.; Verdier, J.; Zuber, H.; Ott, T.; et al. A gene expression atlas of the model legume Medicago truncatula. Plant J. 2008, 55, 504–513. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, R.; Wilson, E. The Theory of Island Biogeography; Princeton University Press: Princeton, NJ, USA, 2001. [Google Scholar]
- Balazadeh, S.; Schildhauer, J.; Araújo, W.L.; Munné-Bosch, S.; Fernie, A.R.; Proost, S.; Humbeck, K.; Mueller-Roeber, B. Reversal of senescence by N resupply to N-starved Arabidopsis thaliana: Transcriptomic and metabolomic consequences. J. Exp. Bot. 2014, 65, 3975–3992. [Google Scholar] [CrossRef] [PubMed]
- Salon, C.; Munier-Jolain, N.; Duc, G.; Voisin, A.-S.; Grandgirard, D.; Larmure, A.; Emery, R.; Ney, B. Grain legume seed filling in relation to nitrogen acquisition: A review and prospects with particular reference to pea. Agronomie 2001, 21, 539–552. [Google Scholar] [CrossRef]
- Zhukov, V.A.; Akhtemova, G.A.; Zhernakov, A.I.; Sulima, A.S.; Shtark, O.Y.; Tikhonovich, I.A. Evaluation of the symbiotic effectiveness of Pea (Pisum Sativum L.) Genotypes in pot experiment. Agric. Biol. 2017, 52, 607–614. [Google Scholar] [CrossRef]
- Osmolovskaya, N.; Shumilina, J.; Kim, A.; Didio, A.; Grishina, T.; Bilova, T.; Keltsieva, O.A.; Zhukov, V.; Tikhonovich, I.; Tarakhovskaya, E.; et al. Methodology of Drought Stress Research: Experimental Setup and Physiological Characterization. Int. J. Mol. Sci. 2018, 19, 4089. [Google Scholar] [CrossRef]
- Kozlova, N.; Strunnikova, O.K. Production and specificity of polyclonal antibodies against soluble proteins from the arbuscular mycorrhizal fungus Glomus intraradices. Mycorrhiza 2001, 10, 301–305. [Google Scholar] [CrossRef]
- Afonin, A.; Sulima, A.; Zhernakov, A.; Zhukov, V. Draft genome of the strain RCAM1026 Rhizobium leguminosarum bv. viciae. Genomics Data 2017, 11, 85–86. [Google Scholar] [CrossRef]
- Shtark, O.Y.; Sulima, A.S.; Zhernakov, A.I.; Kliukova, M.S.; Fedorina, J.V.; Pinaev, A.G.; Kryukov, A.A.; Akhtemova, G.A.; Tikhonovich, I.A.; Zhukov, V.A. Arbuscular mycorrhiza development in pea (Pisum sativum L.) mutants impaired in five early nodulation genes including putative orthologs of NSP1 and NSP2. Symbiosis 2016, 68, 129–144. [Google Scholar] [CrossRef]
- Frolov, A.; Didio, A.; Ihling, C.; Chantzeva, V.; Grishina, T.; Hoehenwarter, W.; Sinz, A.; Smolikova, G.; Bilova, T.; Medvedev, S. The effect of simulated microgravity on the Brassica napus seedling proteome. Funct. Plant Biol. 2018, 45, 440. [Google Scholar] [CrossRef]
- Schmidt, R.; Böhme, D.; Singer, D.; Frolov, A. Specific tandem mass spectrometric detection of AGE-modified arginine residues in peptides. J. Mass Spectrom. 2015, 50, 613–624. [Google Scholar] [CrossRef]
- Greifenhagen, U.; Frolov, A.; Blüher, M.; Hoffmann, R. Plasma Proteins Modified by Advanced Glycation End Products (AGEs) Reveal Site-specific Susceptibilities to Glycemic Control in Patients with Type 2 Diabetes. J. Biol. Chem. 2016, 291, 9610–9616. [Google Scholar] [CrossRef]
- Frolov, A.; Bilova, T.; Paudel, G.; Berger, R.; Balcke, G.U.; Birkemeyer, C.; Wessjohann, L.A. Early responses of mature Arabidopsis thaliana plants to reduced water potential in the agar-based polyethylene glycol infusion drought model. J. Plant Physiol. 2017, 208, 70–83. [Google Scholar] [CrossRef]
- Spiller, S.; Frolov, A.; Hoffmann, R. Quantification of Specific Glycation Sites in Human Serum Albumin as Prospective Type 2 Diabetes Mellitus Biomarkers. Protein Pept. Lett. 2018, 24. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Côté, R.G.; Csordas, A.; Dianes, J.A.; Fabregat, A.; Foster, J.M.; Griss, J.; Alpi, E.; Birim, M.; Contell, J.; et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: Status in 2013. Nucleic Acids Res. 2013, 41, D1063–D1069. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef] [PubMed]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Data files are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clusters of Protein a (8274/3358) | Nr. | Proteins | log2 Fold Change e | Anovap f | qg | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Accession | Description b | Function d | 8274 | 3358 | BSM | NI | |||||
| 1. Up/Up | |||||||||||
 | 1+ | Q1S053 | Probable histone H2A.3 | Chromatin organization | 1.5 | NS | NS | 1.3 | 0.012 | 0.032 | |
| 2+ | I3SCW0 | Uncharacterized; HSP20-like chaperone c | External stimuli response | 2.7 | 1.7 | NS | NS | 0.023 | 0.043 | ||
| 3 | A0A072UBI6 | Small hydrophilic plant seed protein | Not assigned | 1.7 | 1.2 | 2.3 | 1.6 | 0.016 | 0.038 | ||
| 4 | Lj0g3v0065729.1 | Uncharacterized; 60S ribosomal protein L35-like c | Protein biosynthesis | 1.1 | 0.6 | NS | 1.6 | 0.011 | 0.029 | ||
| 2. Up/Down | |||||||||||
 | 2.1 | 5+ | Medtr1g102870.1 | 1,2-dihydroxy-3-keto-5-methylthiopentene dioxygenase | Amino acid metabolism | 1.9 | −1.3 | NS | NS | 0.011 | 0.032 |
| 6 | A0A072VC98 | ATPase. AAA-type. CDC48 protein | Not assigned | 1.0 | −0.6 | 3.5 | 3.2 | 0.024 | 0.044 | ||
| 7 | I3T832 | Uncharacterized; response to oxidative stress, heme binding, peroxidase activity c | 1.6 | −0.7 | −0.8 | 1.0 | 0.005 | 0.018 | |||
| 8 | Lj6g3v1880130.1 | ATP-dependent (S)-NAD(P)H-hydrate dehydratase | 1.3 | −1.0 | −0.6 | 1.3 | 0.023 | 0.044 | |||
| 9 | A0A072U0B5 | UDP-glucosyltransferase family protein | Other enzyme families | 0.9 | −0.8 | 2.0 | 1.9 | 0.012 | 0.031 | ||
| 10 | G7IEE7 | Xyloglucanase-specific endoglucanase inhibitor p. | Protein degradation | 1.0 | NS | −4.6 | −4.5 | 0.002 | 0.014 | ||
| 11+ | B7FH22 | Uncharacterized; oxidoreductase activity c | Redox homeostasis | 2.3 | −0.7 | NS | 0.8 | 0.024 | 0.043 | ||
| 12+ | G7L8T3 | Guanosine nucleotide diphosphate dissociation inhibitor | Vesicle trafficking | 1.7 | −1.7 | −5.1 | −5.2 | 0.020 | 0.043 | ||
| 2.2 | 13 | G7IJ13 | Proteasome subunit alpha type | Protein degradation | 1.3 | −0.7 | −2.3 | −2.1 | 0.025 | 0.044 | |
| 14 | B7FLD1 | Putative uncharacterized; Nop domain superfamily (pre-RNA processing ribonucleoproteins) c | RNA processing | 1.8 | −1.8 | 0.6 | NS | 0.001 | 0.013 | ||
| 15 | A0A072UGB7 | Clathrin heavy chain | Vesicle trafficking | 1.4 | −1.4 | 3.7 | 2.6 | 0.034 | 0.050 | ||
| 3. Up/Steady | |||||||||||
 | 3.1 | 16 | A0A072W1H5 | ATP synthase subunit beta | Cellular respiration | 3.0 | NS | NS | 2.7 | 0.003 | 0.017 |
| 17 | I3SN66 | Uncharacterized; triose-phosphate isomerase activity, chloroplast organization c | 2.7 | 1.0 | −0.6 | −0.8 | 0.004 | 0.017 | |||
| 18 | G7IUE0 | LRR receptor-like kinase family protein | Not assigned | 2.1 | NS | NS | NS | 0.010 | 0.029 | ||
| 19 | G7J538 | GDP-fucose protein O-fucosyltransferase | 1.6 | NS | −0.7 | −0.9 | 0.023 | 0.044 | |||
| 20 | V7CPQ1 | Uncharacterized; ATP binding c | 1.8 | NS | 0.8 | 2.9 | 0.002 | 0.015 | |||
| 21+ | A2Q582 | Aldo/keto reductase | Other enzyme families | 1.9 | NS | NS | 3.3 | 0.016 | 0.038 | ||
| 22 | A0A072UJ10 | Cytoplasmic ribosomal protein S13 | Protein biosynthesis | 3.9 | 0.8 | 2.6 | 2.5 | 0.002 | 0.015 | ||
| 23 | I3T617 | 60S ribosomal L35-like protein | 4.0 | 1.4 | NS | 2.4 | 0.005 | 0.018 | |||
| 24+ | Q5QQ34 | Coatomer epsilon subunit | Vesicle trafficking | 2.8 | NS | NS | 1.5 | 0.019 | 0.041 | ||
| 3.2 | 25 | I3SHC8 | Uncharacterized; ribosome biogenesis c | Protein biosynthesis | 1.7 | NS | NS | −0.6 | 0.008 | 0.025 | |
 | 4.1 | 26 | V7BQ03 | Uncharacterized; carboxy-lyase activity, magnesium ion binding, thiamine pyrophosphate binding c | Carbohydrate metabolism | NS | NS | −2.9 | 0.8 | 0.037 | 0.050 |
| 27 | G7KBA2 | 17.6 kDa class I heat shock protein | External stimuli response | NS | 1.0 | −1.2 | −1.3 | 0.003 | 0.015 | ||
| 28 | A0A072TF91 | Heat shock protein HSP20. putative (Fragment) | Not assigned | NS | NS | 2.5 | 1.8 | 0.030 | 0.046 | ||
| 29 | Lj3g3v0324640.1 | Lipoxygenase | Other enzyme families | NS | NS | −1.7 | 1.1 | 0.004 | 0.018 | ||
| 30+ | V7BZK0 | Lipoxygenase | NS | NS | 2.1 | 2.9 | 0.004 | 0.019 | |||
| 4.2 | 31 | G7L0I7 | Cobalamin-independent methionine synthase | Amino acid metabolism | NS | NS | −1.1 | −5.0 | 0.012 | 0.031 | |
| 32 | G7L831 | TCP-1/cpn60 chaperonin family protein | Cytoskeleton | NS | 0.9 | −0.7 | −1.7 | 0.023 | 0.044 | ||
| 33 | G7JSC7 | NB-ARC domain disease resistance protein | Not assigned | 0.6 | 0.6 | −1.2 | −1.3 | 0.002 | 0.014 | ||
| 34 | Lj1g3v0411500.1 | Uncharacterized; Myb/SANT-like domain (nuclear DNA-binding proteins, nuclear receptor co-repressors) c | NS | 0.6 | NS | 1.8 | 0.016 | 0.038 | |||
| 35 | V7ARA2 | Uncharacterized; Ca2+ binding c | NS | NS | 1.6 | 2.1 | 0.001 | 0.012 | |||
| 36 | V7BSM8 | Annexin | NS | NS | 0.6 | −0.8 | 0.001 | 0.013 | |||
| 37 | V7AR99 | Uncharacterized; lipase activity c | NS | 1.0 | 1.7 | 2.6 | 0.016 | 0.038 | |||
| 38 | B7FIG5 | Putative uncharacterized; oxidoreductase activity, acting on the CH–CH group of donors c | Nucleotide metabolism | NS | 0.7 | 1.5 | 2.6 | 0.001 | 0.013 | ||
| 39 | A0A072V122 | tRNA-binding region domain protein | Protein biosynthesis | NS | NS | 2.4 | 1.9 | 0.024 | 0.044 | ||
| 40 | V7AUC2 | Uncharacterized; RNA 3′-end processing, RNA polyadenylation c | RNA processing | NS | NS | −0.6 | NS | 0.013 | 0.032 | ||
| 41+ | I3SYE6 | 1,2-dihydroxy-3-keto-5-methylthiopentene dioxygenase | Amino acid metabolism | NS | NS | NS | -0.6 | 0.009 | 0.027 | ||
 | 42 | G7J834 | Glucose-1-phosphate adenylyltransferase | Carbohydrate metabolism | NS | NS | −0.9 | −0.6 | 0.024 | 0.044 | |
| 43 | G7JM88 | Lethal leaf-spot protein. putative | Coenzyme metabolism | NS | −1.1 | −0.6 | −1.1 | 0.018 | 0.040 | ||
| 44 | Lj4g3v2371610.1 | Probable glycine cleavage T-protein family (aminomethyl transferase) | −0.9 | NS | −2.1 | 1.5 | 0.004 | 0.017 | |||
| 45+ | Medtr5g019780.1 | Cupin family protein | Not assigned | NS | −1.0 | NS | 1.1 | 0.013 | 0.033 | ||
| 46 | A0A072TDJ4 | TCP-1/cpn60 chaperonin family protein (Fragment) | −1.4 | −1.1 | 1.4 | 1.2 | 0.037 | 0.051 | |||
| 47 | G7IDU4 | Protein disulfide isomerase-like protein | −1.1 | −1.0 | −1.6 | −1.4 | 0.003 | 0.017 | |||
| 48 | V7AJE4 | Probable defense response, ADP binding | NS | −2.0 | 1.8 | 1.7 | 0.024 | 0.044 | |||
| 49+ | B7FJF0 | Xylose isomerase | Other enzyme families | −2.7 | −3.5 | NS | 1.6 | 0.021 | 0.043 | ||
| 50+ | G7ILF2 | 60S ribosomal protein L26-1 | Protein biosynthesis | −0.6 | NS | −1.0 | −1.2 | 0.031 | 0.047 | ||
| 51+ | I3T560 | Superoxide dismutase | Redox homeostasis | NS | −0.8 | −1.4 | 1.7 | 0.023 | 0.043 | ||
| 6. Down/Steady | |||||||||||
 | 6.1 | 52+ | I3SU69 | Uncharacterized; argininosuccinatelyase activity c | Amino acid metabolism | NS | NS | −0.9 | −1.5 | 0.001 | 0.010 |
| 53 | G7J530 | Argininosuccinatelyase | NS | NS | −0.6 | −0.8 | 0.001 | 0.009 | |||
| 54+ | A0A072TSD1 | Pectin acetylesterase | Cell wall | −0.9 | NS | 1.3 | 1.2 | 0.008 | 0.026 | ||
| 55 | G7JFK1 | Heat shock 70 kDa protein | External stimuli response | NS | 0.8 | −0.7 | −0.7 | 0.001 | 0.010 | ||
| 56+ | V7C9P5 | Uncharacterized; ATP binding c | NS | 0.8 | 3.5 | 3.2 | 0.001 | 0.010 | |||
| 57 | A0A072U2T6 | Translin-like protein | Not assigned | NS | NS | 0.9 | 0.9 | 0.000 | 0.008 | ||
| 58+ | A0A072TMR3 | Late embryogenesis abundant protein | −1.3 | NS | 1.2 | 0.7 | 0.000 | 0.001 | |||
| 59+ | B1NY79 | Cold-acclimation specific protein 15 | NS | 0.9 | −0.7 | 1.3 | 0.005 | 0.020 | |||
| 60+ | B5U8K3 | Convicilin storage protein 1 | NS | NS | NS | 1.2 | 0.001 | 0.010 | |||
| 61+ | I3S2D8 | Uncharacterized; Mitochondrial inner membrane translocase subunit c | 2.4 | NS | −0.6 | −1.4 | 0.000 | 0.000 | |||
| 62+ | V7BVA1 | Uncharacterized; QWRF domain family, microtubule-associated c | −0.6 | −0.6 | −1.3 | NS | 0.000 | 0.000 | |||
| 63 | I3T8A0 | Glutamine synthetase | Nutrient uptake | NS | NS | NS | 1.4 | 0.025 | 0.044 | ||
| 64 | G7IS29 | Lipoxygenase | Other enzyme families | NS | NS | NS | −0.8 | 0.024 | 0.044 | ||
| 65+ | Medtr1g094155.1 | Probable serine carboxypeptidase-like protein | Protein degradation | −1.0 | −0.6 | NS | −0.6 | 0.023 | 0.043 | ||
| 66 | G7I549 | 26S proteasome non-ATPase regulatory subunit-like protein | NS | NS | 0.7 | 0.6 | 0.034 | 0.050 | |||
| 67 | A0A072TQN5 | Phosphatase 2C family protein | Protein modification | NS | NS | 0.9 | 0.6 | 0.003 | 0.016 | ||
| 68 | B7FMC4 | Putative uncharacterized; Glutathione S-transferases terminal domain c | −0.6 | NS | 1.6 | NS | 0.023 | 0.044 | |||
| 69 | G7LH03 | Glycosyltransferase | Secondary metabolism | NS | NS | −2.1 | −2.7 | 0.000 | 0.008 | ||
| 6.2 | 70 | G7JPY4 | Delta-1-pyrroline-5-carboxylate dehydrogenase | Amino acid metabolism | NS | NS | −0.9 | −0.7 | 0.004 | 0.017 | |
| 71 | A0A072VNG1 | Uncharacterized protein | Not assigned | NS | NS | NS | 2.1 | 0.002 | 0.014 | ||
| 72 | Lj1g3v3690420.1 | Elongation factor 1-alpha | Protein biosynthesis | NS | NS | NS | 2.7 | 0.026 | 0.044 | ||
| 73 | G7IVL9 | U-box kinase family protein | Protein modification | NS | NS | NS | −0.8 | 0.034 | 0.050 | ||
| 74 | Lj0g3v0348019.1 | Transcription factor | RNA biosynthesis | NS | NS | 0.8 | 2.9 | 0.002 | 0.014 | ||
| 75 | Q93XA4 | Homeodomain leucine zipper protein HDZ2 | NS | NS | −1.1 | NS | 0.007 | 0.022 | |||
| 7. Steady/Up | |||||||||||
 | 76 | V7AU77 | Uncharacterized; lactoylglutathionelyase activity c | Cellular respiration | 0.8 | NS | −0.9 | NS | 0.030 | 0.046 | |
| 77 | G7IHB8 | Core histone H2A/H2B/H3/H4 | Chromatin organization | 0.8 | NS | −0.7 | −0.7 | 0.025 | 0.044 | ||
| 78+ | Q38JC8 | Temperature-induced lipocalin | Not assigned | 1.0 | 1.7 | 1.1 | 0.6 | 0.015 | 0.036 | ||
| 79 | G7JPM2 | Uro-adherence factor A. putative | NS | 0.9 | −1.0 | −1.0 | 0.027 | 0.046 | |||
| 80 | V7ALP7 | Annexin | 0.6 | 0.6 | 1.7 | 1.9 | 0.013 | 0.032 | |||
| 81 | B7FJY0 | Annexin | 0.8 | 0.6 | −0.9 | 3.6 | 0.010 | 0.029 | |||
| 82 | V7B712 | Hexosyltransferase | 0.9 | 1.0 | −0.9 | NS | 0.035 | 0.050 | |||
| 83 | V7BYE1 | Uncharacterized (Fragment); Leucine-rich repeat domain superfamily c | 0.9 | 1.4 | −1.4 | 0.6 | 0.037 | 0.050 | |||
| 84+ | A0A072U7T5 | F-box/RNI/F box domain-like domain protein | 0.7 | 4.6 | 6.9 | 5.8 | 0.003 | 0.017 | |||
| 85 | B7FK47 | Ferritin | Nutrient uptake | 0.7 | 0.9 | 2.4 | 3.3 | 0.031 | 0.048 | ||
| 86 | A0A072UUP4 | 60S ribosomal protein L18a | Protein biosynthesis | NS | 0.9 | 1.4 | 1.1 | 0.029 | 0.046 | ||
| 87 | A0A072VAP6 | 60S ribosomal protein L17A | 0.6 | NS | 8.9 | 8.9 | 0.032 | 0.049 | |||
| 88 | A0A072VJE7 | 40S ribosomal protein S5-2 | NS | 0.9 | 0.7 | 2.4 | 0.028 | 0.046 | |||
| 89+ | G7IH13 | Translation elongation factor EF-2 subunit | 0.6 | 1.7 | −1.7 | −1.8 | 0.025 | 0.043 | |||
| 90 | B7FMQ6 | 60S ribosomal L23-like protein | 0.7 | 0.9 | 0.7 | 0.6 | 0.036 | 0.050 | |||
| 91 | V7B0F4 | Uncharacterized; RNA-binding, RNA-mediated gene silencing c | RNA biosynthesis | 0.8 | 1.0 | −1.8 | −0.8 | 0.006 | 0.021 | ||
| 92+ | I3SRR2 | Uncharacterized; transcription factor activity, sequence-specific DNA binding, zinc ion binding c | 0.7 | 0.9 | −0.6 | −0.8 | 0.011 | 0.032 | |||
| 93 | G7J2R6 | 110 kDa 4SNc-tudor domain protein | RNA processing | NS | NS | −1.1 | −0.8 | 0.008 | 0.025 | ||
| 8. Steady/Down | |||||||||||
 | 8.1 | 94 | V7AWC5 | 4-alpha-glucanotransferase | Carbohydrate metabolism | NS | NS | −1.4 | NS | 0.033 | 0.049 |
| 95 | G9JLT6 | ATP synthase subunit alpha | Cellular respiration | NS | −0.8 | −0.7 | −0.9 | 0.004 | 0.017 | ||
| 96+ | G7KG34 | Glutamine synthetase | Nutrient uptake | NS | NS | −0.7 | −1.2 | 0.003 | 0.016 | ||
| 97 | G7IH71 | Phosphoenolpyruvate carboxylase | Photosynthesis | NS | −0.6 | −1.2 | −1.6 | 0.000 | 0.008 | ||
| 98 | G7IBY1 | 60S ribosomal protein L3B | Protein biosynthesis | 0.7 | −1.3 | −1.3 | −1.2 | 0.003 | 0.017 | ||
| 99+ | B7FN14 | Uncharacterized; Involved in translation, rRNA-binding c | 0.7 | −3.8 | 1.0 | 0.8 | 0.002 | 0.014 | |||
| 100+ | A0A072TQ47 | Phosphatase 2C family protein | Protein modification | 0.6 | NS | NS | 0.8 | 0.015 | 0.036 | ||
| 101+ | A0PG70 | Catalase | Redox homeostasis | NS | −1.4 | NS | 2.7 | 0.021 | 0.043 | ||
| 8.2 | 102 | A0A072UKG0 | Histone H2B | Chromatin organization | 0.8 | NS | 11.4 | 13.5 | 0.005 | 0.018 | |
| 103 | V7B8C8 | Uncharacterized; translation, structural constituent of ribosome c | Protein biosynthesis | 0.8 | −1.1 | −2.1 | NS | 0.009 | 0.026 | ||
| 104+ | A0A072VE37 | UTP-glucose-1-phosphate uridylyltransferase | 1.5 | −2.1 | NS | 2.2 | 0.008 | 0.026 | |||
| 8.3 | 105 | A0A072VJU4 | Glutathione S-transferase. amino-terminal domain protein | Protein modification | NS | −1.4 | 0.7 | 1.9 | 0.005 | 0.018 | |
| 9. Steady/Steady | |||||||||||
 | 9.1 | 106 | A2Q4V2 | Leucine-rich repeat. plant specific | Not assigned | NS | NS | −0.9 | 2.5 | 0.000 | 0.008 |
| 107 | I3SIG9 | Chlorophyll a-b binding protein. chloroplastic | Photosynthesis | 0.6 | NS | NS | −0.9 | 0.000 | 0.000 | ||
| 108 | B6DXD7 | Vacuolar H+-translocating inorganic pyrophosphatase | Solute transport | NS | NS | 0.9 | 2.1 | 0.008 | 0.025 | ||
| 9.2 | 109+ | G7IMZ3 | ABA-responsive protein | Not assigned | 1.4 | NS | −1.6 | −1.9 | 0.003 | 0.017 | |
| 10. None h | |||||||||||
| 110 | A0A072TYG8 | β-hexosaminidase | Protein modification | - | NS | - | - | 0.000 | 0.000 | ||
| 111 | Lj0g3v0098069.1 | Uncharacterized; l-type lectin-domain containing receptor kinase S.7-like c | Not assigned | NS | - | - | - | 0.000 | 0.000 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamontova, T.; Afonin, A.M.; Ihling, C.; Soboleva, A.; Lukasheva, E.; Sulima, A.S.; Shtark, O.Y.; Akhtemova, G.A.; Povydysh, M.N.; Sinz, A.; et al. Profiling of Seed Proteome in Pea (Pisum sativum L.) Lines Characterized with High and Low Responsivity to Combined Inoculation with Nodule Bacteria and Arbuscular Mycorrhizal Fungi. Molecules 2019, 24, 1603. https://doi.org/10.3390/molecules24081603
Mamontova T, Afonin AM, Ihling C, Soboleva A, Lukasheva E, Sulima AS, Shtark OY, Akhtemova GA, Povydysh MN, Sinz A, et al. Profiling of Seed Proteome in Pea (Pisum sativum L.) Lines Characterized with High and Low Responsivity to Combined Inoculation with Nodule Bacteria and Arbuscular Mycorrhizal Fungi. Molecules. 2019; 24(8):1603. https://doi.org/10.3390/molecules24081603
Chicago/Turabian StyleMamontova, Tatiana, Alexey M. Afonin, Christian Ihling, Alena Soboleva, Elena Lukasheva, Anton S. Sulima, Oksana Y. Shtark, Gulnara A. Akhtemova, Maria N. Povydysh, Andrea Sinz, and et al. 2019. "Profiling of Seed Proteome in Pea (Pisum sativum L.) Lines Characterized with High and Low Responsivity to Combined Inoculation with Nodule Bacteria and Arbuscular Mycorrhizal Fungi" Molecules 24, no. 8: 1603. https://doi.org/10.3390/molecules24081603
APA StyleMamontova, T., Afonin, A. M., Ihling, C., Soboleva, A., Lukasheva, E., Sulima, A. S., Shtark, O. Y., Akhtemova, G. A., Povydysh, M. N., Sinz, A., Frolov, A., Zhukov, V. A., & Tikhonovich, I. A. (2019). Profiling of Seed Proteome in Pea (Pisum sativum L.) Lines Characterized with High and Low Responsivity to Combined Inoculation with Nodule Bacteria and Arbuscular Mycorrhizal Fungi. Molecules, 24(8), 1603. https://doi.org/10.3390/molecules24081603