Recent Advances in HIV-1 Gag Inhibitor Design and Development


Abstract
:1. Introduction and Current Status of Antiretroviral Therapies
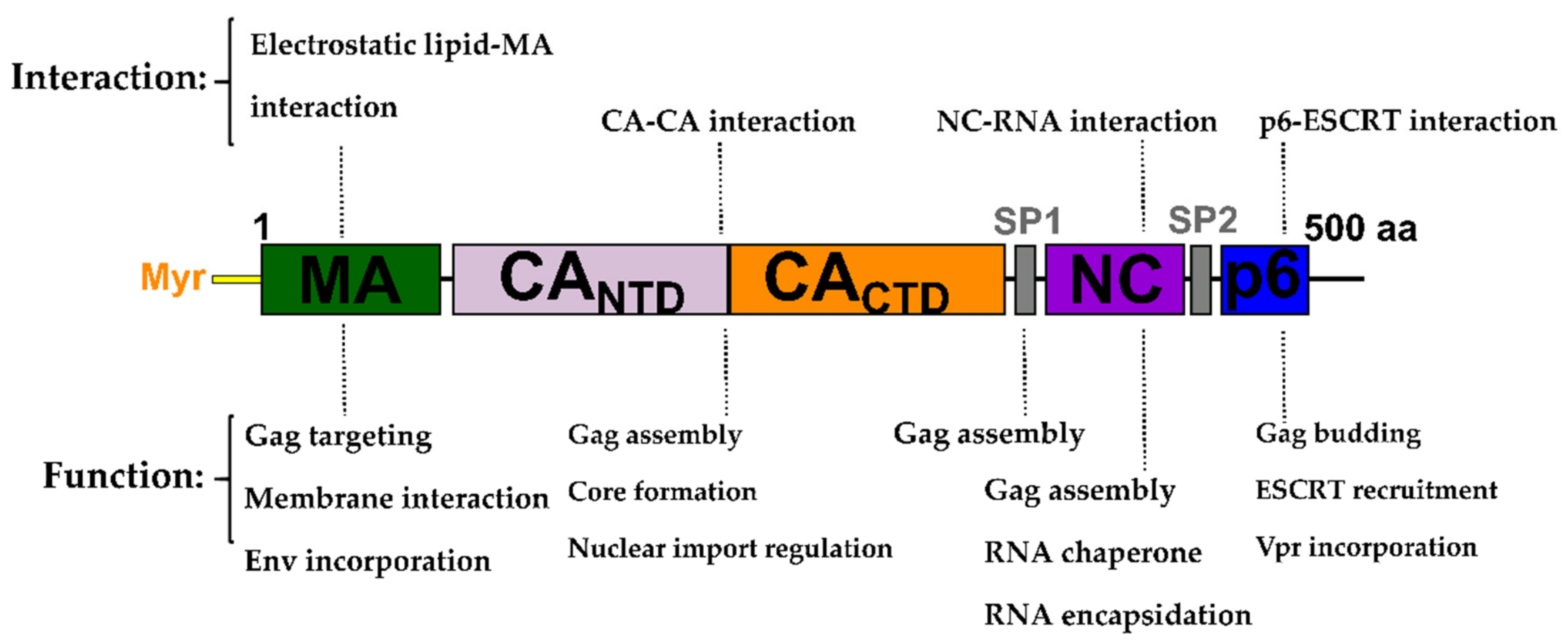
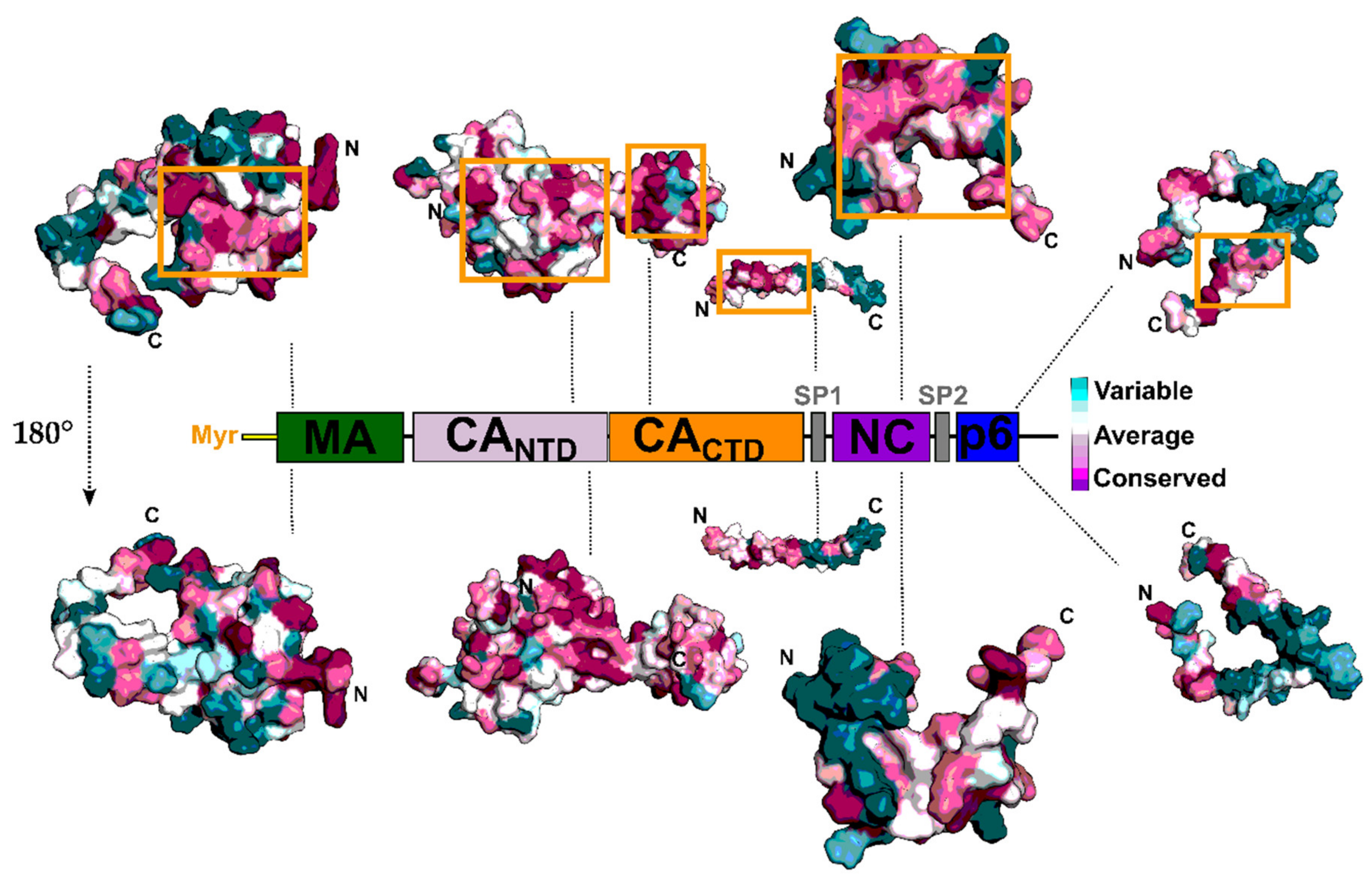
2. The Gag Polyprotein and Its Role in the HIV-1 Replication Cycle
3. HIV-1 Protease and Maturation Inhibitors
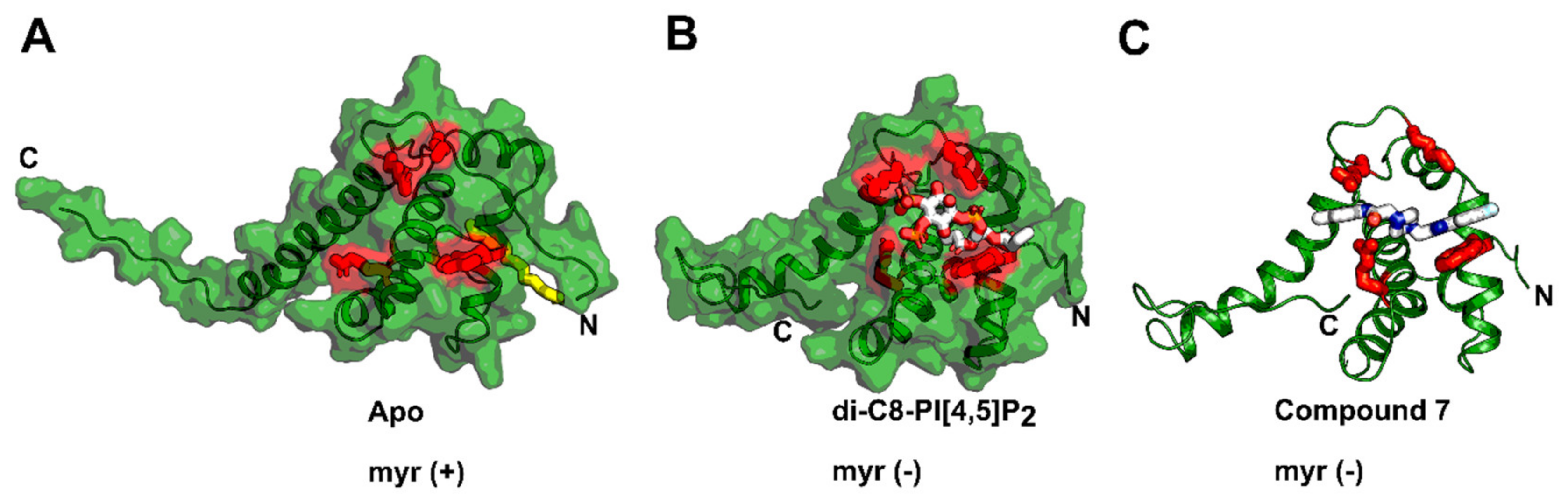
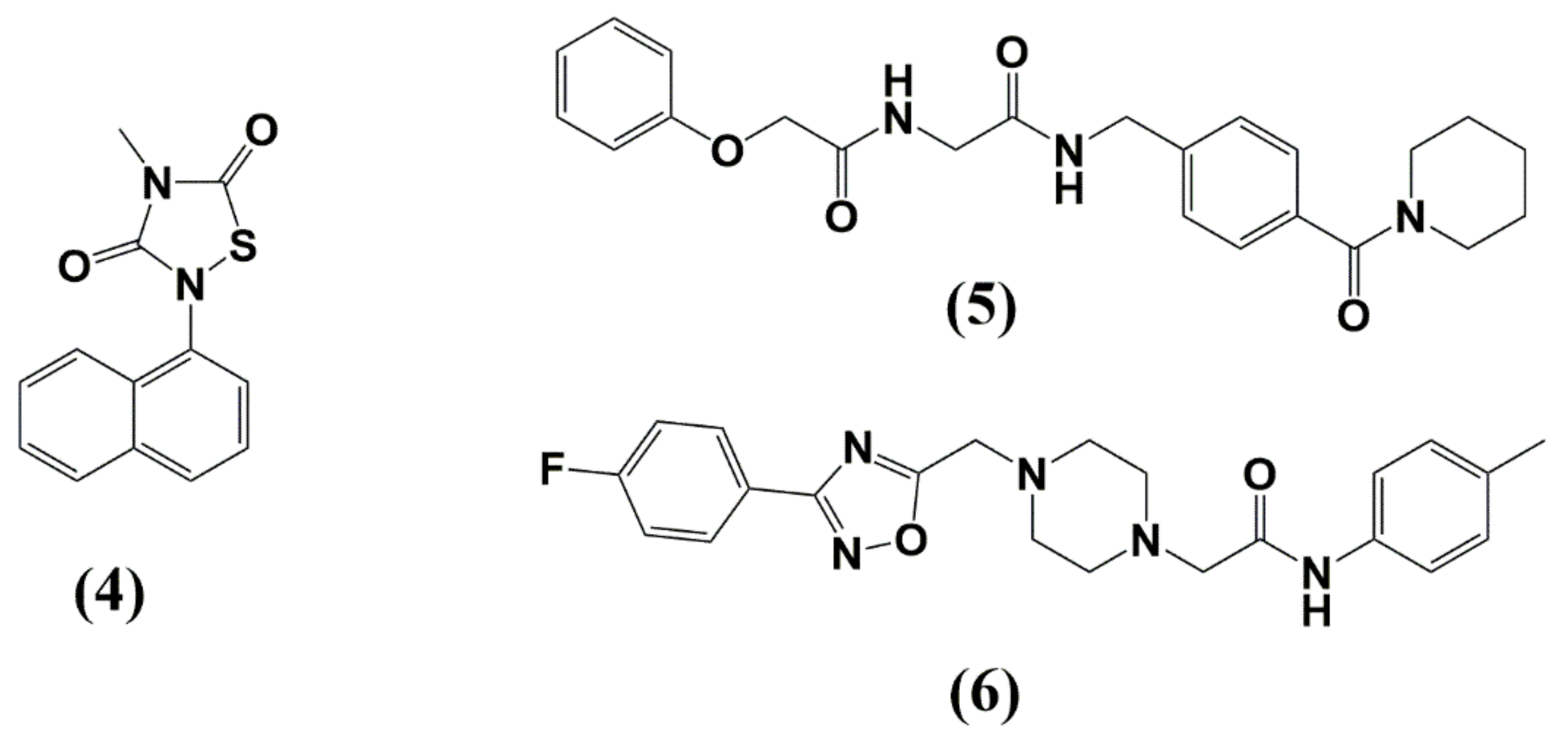
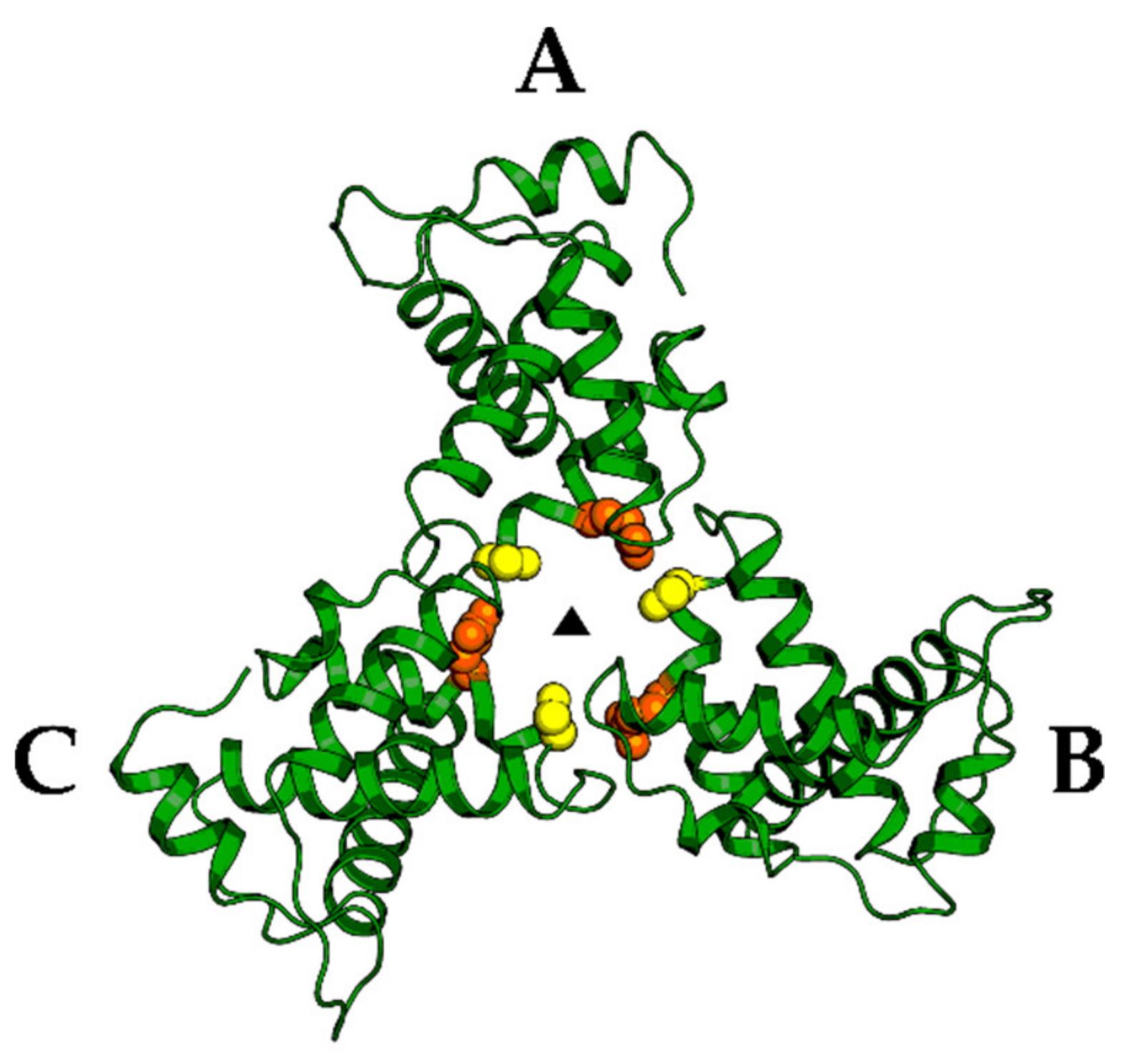
4. Matrix (MA, p17)
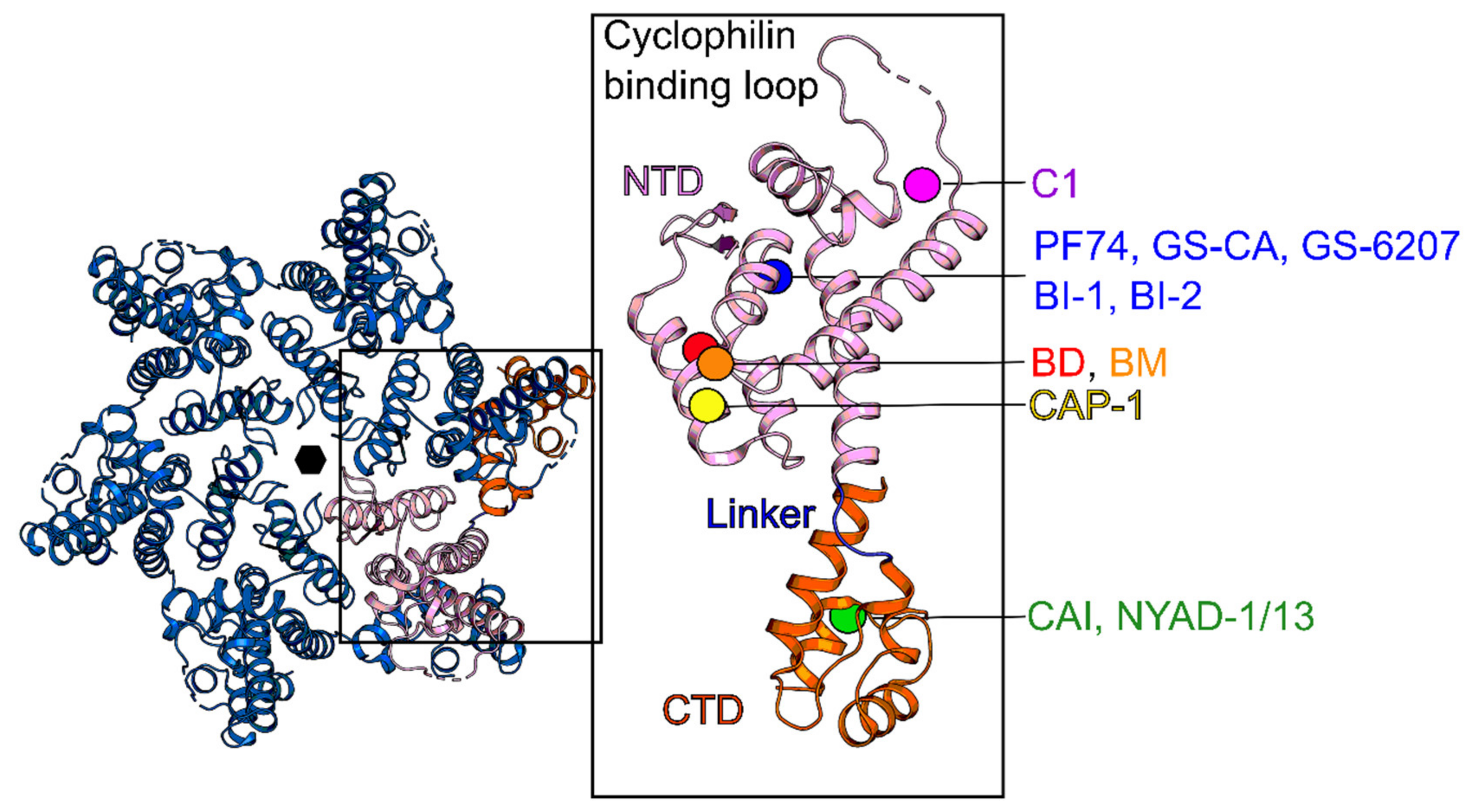
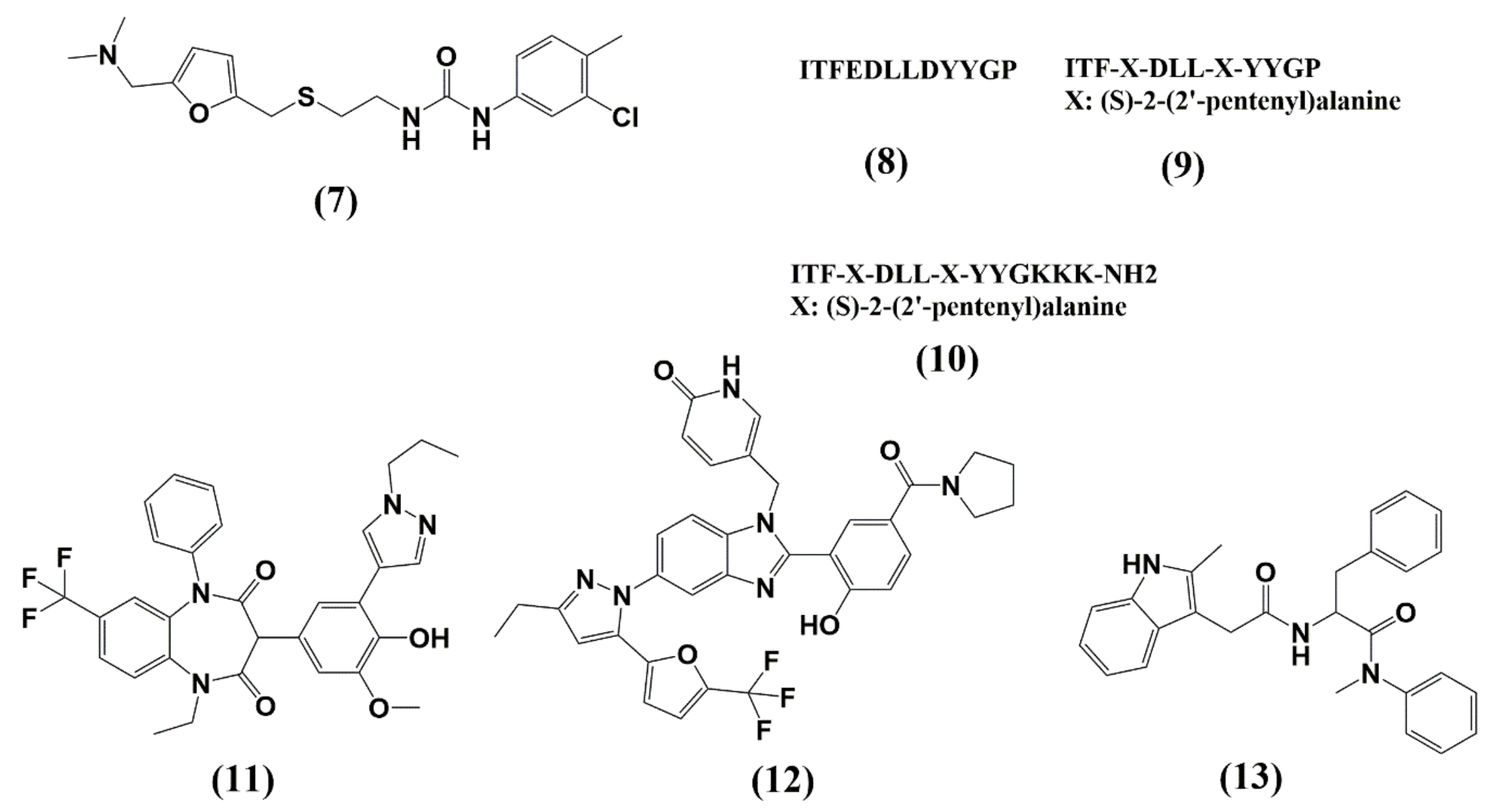
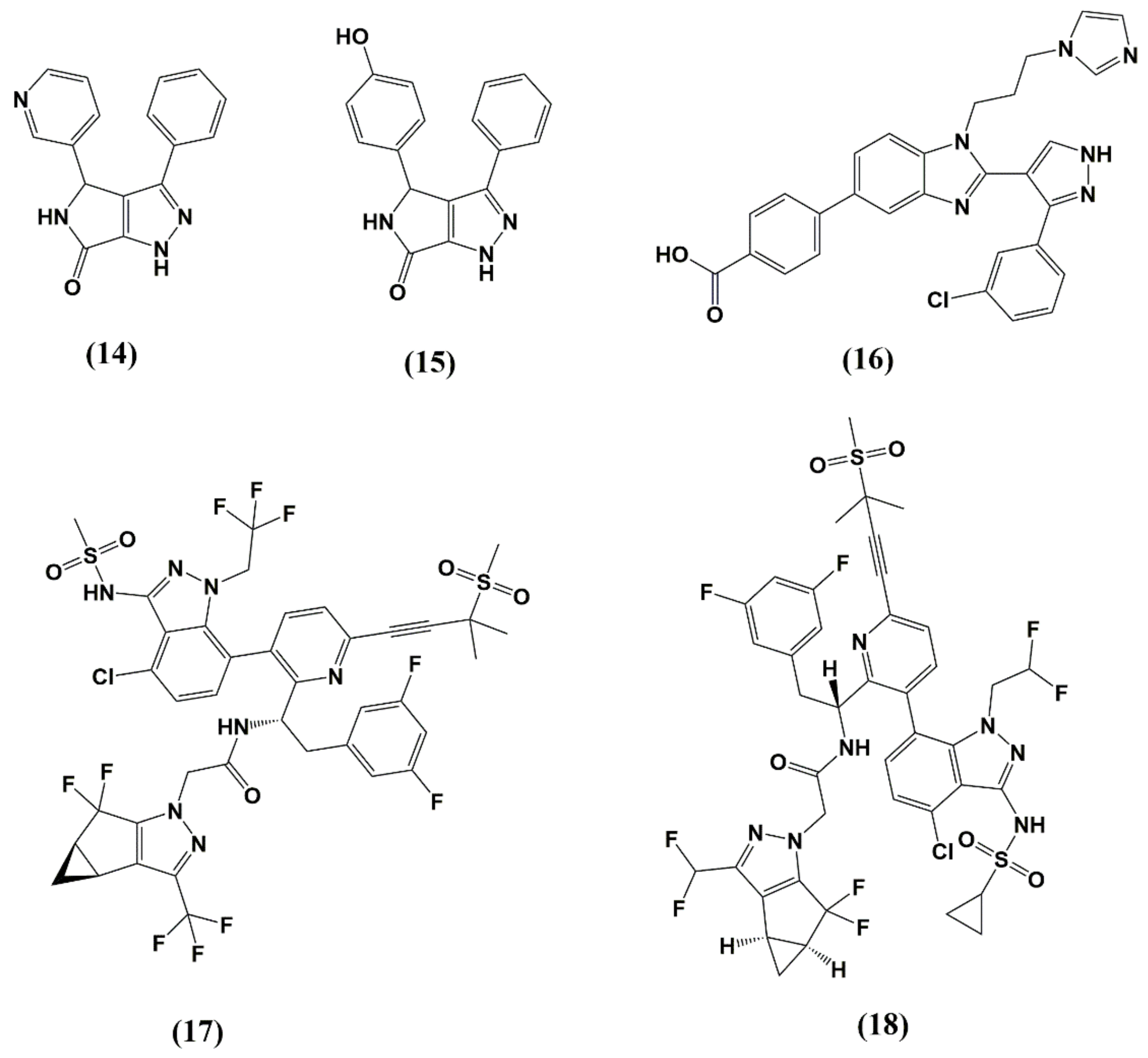
5. Capsid (CA, p24)
6. Nucleocapsid (NC, NCp7)
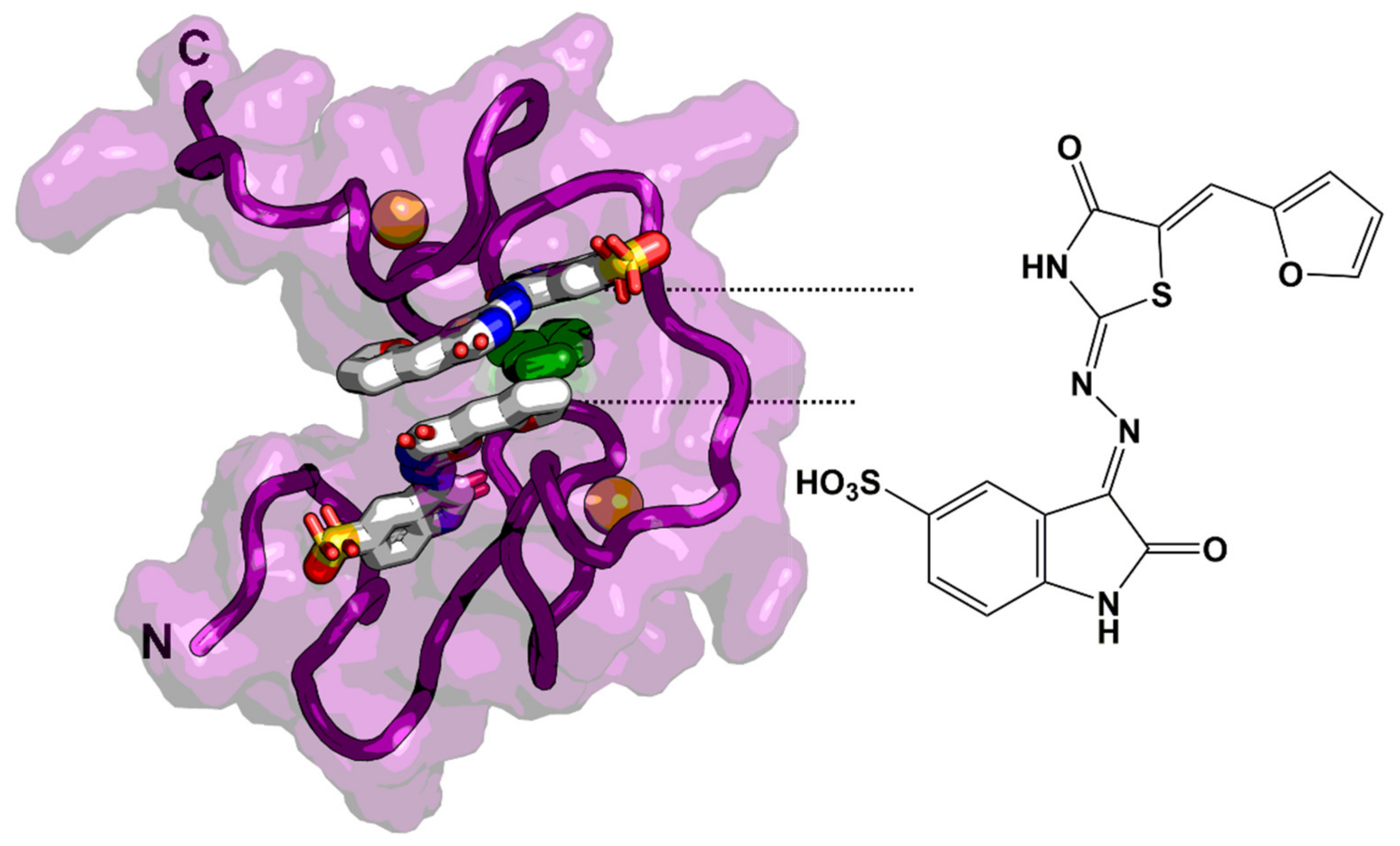
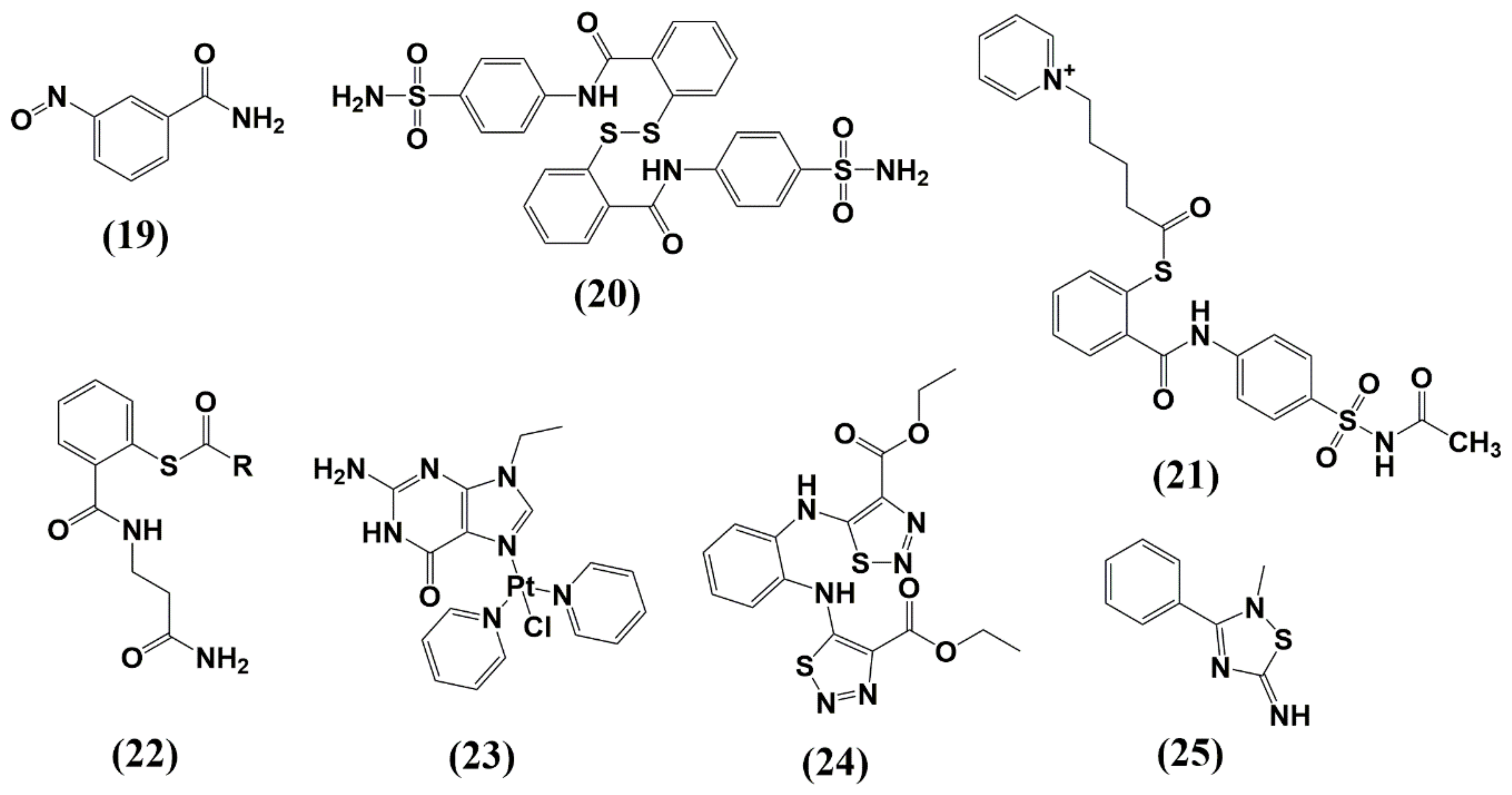
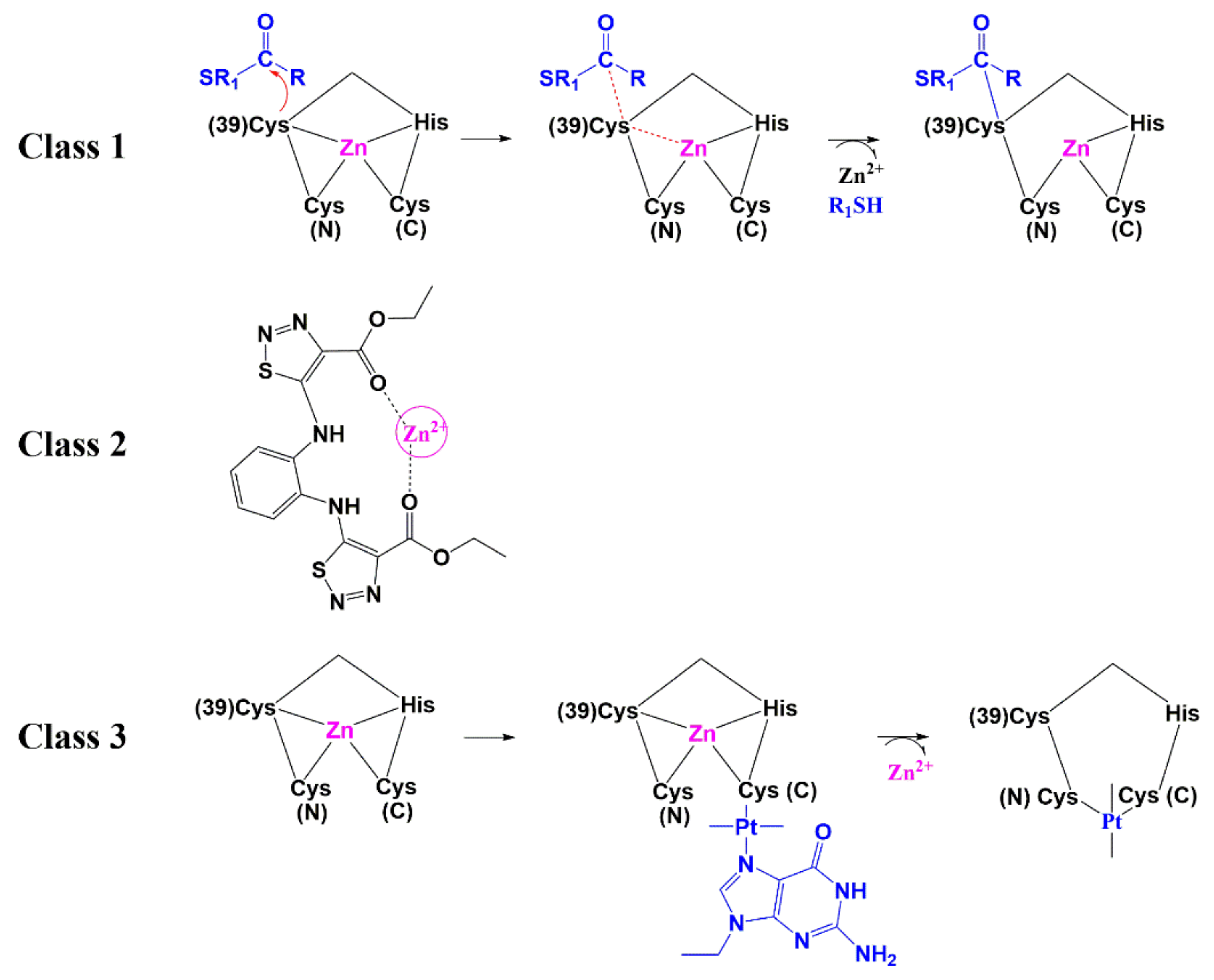
6.1. Zinc-Ejectors
6.2. Small Molecules as Non-Zinc Ejectors
7. Late Domains (P6)
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef] [Green Version]
- UNAIDS. Global HIV & AIDS Statistics—2019 Fact Sheet, United Nations. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 1 February 2020).
- Zhang, X. Anti-retroviral drugs: Current state and development in the next decade. Acta Pharm. Sin. B 2018, 8, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef] [PubMed]
- Cihlar, T.; Fordyce, M. Current status and prospects of HIV treatment. Curr. Opin. Virol. 2016, 18, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, S. Advances in detection and monitoring of plasma viremia in HIV-infected individuals receiving antiretroviral therapy. Curr. Opin. HIV AIDS 2013, 8, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, F.; Lodwick, R.K.; Smith, C.J.; Smith, R.; Cambiano, V.; Lundgren, J.D.; Delpech, V.; Phillips, A.N. Projected life expectancy of people with HIV according to timing of diagnosis. AIDS 2012, 26, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Guaraldi, G.; Orlando, G.; Zona, S.; Menozzi, M.; Carli, F.; Garlassi, E.; Berti, A.; Rossi, E.; Roverato, A.; Palella, F. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin. Infect. Dis. 2011, 53, 1120–1126. [Google Scholar] [CrossRef] [Green Version]
- Freed, E.O. HIV-1 assembly, release and maturation, Nature reviews. Microbiology 2015, 13, 484–496. [Google Scholar]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Chukkapalli, V.; Ono, A. Molecular determinants that regulate plasma membrane association of HIV-1 Gag. J. Mol. Biol. 2011, 410, 512–524. [Google Scholar] [CrossRef] [Green Version]
- Saad, J.S.; Miller, J.; Tai, J.; Kim, A.; Ghanam, R.H.; Summers, M.F. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. USA 2006, 103, 11364–11369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed, E.O.; Martin, M.A. Virion incorporation of envelope glycoproteins with long but not short cytoplasmic tails is blocked by specific, single amino acid substitutions in the human immunodeficiency virus type 1 matrix. J. Virol. 1995, 69, 1984–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.C. Mechanisms for Env glycoprotein acquisition by retroviruses. AIDS Res. Hum. Retrovir. 2011, 27, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundquist, W.I.; Krausslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- Novikova, M.; Adams, L.J.; Fontana, J.; Gres, A.T.; Balasubramaniam, M.; Winkler, D.C.; Kudchodkar, S.B.; Soheilian, F.; Sarafianos, S.G.; Steven, A.C.; et al. Identification of a Structural Element in HIV-1 Gag Required for Virus Particle Assembly and Maturation. mBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Matreyek, K.A.; Engelman, A. Viral and cellular requirements for the nuclear entry of retroviral preintegration nucleoprotein complexes. Viruses 2013, 5, 2483–2511. [Google Scholar] [CrossRef] [Green Version]
- Muriaux, D.; Darlix, J.L. Properties and functions of the nucleocapsid protein in virus assembly. RNA Biol. 2010, 7, 744–753. [Google Scholar] [CrossRef]
- Lu, K.; Heng, X.; Summers, M.F. Structural determinants and mechanism of HIV-1 genome packaging. J. Mol. Biol. 2011, 410, 609–633. [Google Scholar] [CrossRef] [Green Version]
- Rein, A.; Henderson, L.E.; Levin, J.G. Nucleic-acid-chaperone activity of retroviral nucleocapsid proteins: Significance for viral replication. Trends Biochem. Sci. 1998, 23, 297–301. [Google Scholar] [CrossRef]
- Rein, A. Nucleic acid chaperone activity of retroviral Gag proteins. RNA Biol. 2010, 7, 700–705. [Google Scholar] [CrossRef] [Green Version]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.A.; Temeselew, L.G.; Crist, R.M.; Soheilian, F.; Kamata, A.; Mirro, J.; Harvin, D.; Nagashima, K.; Cachau, R.E.; Rein, A. On the role of the SP1 domain in HIV-1 particle assembly: A molecular switch? J. Virol. 2011, 85, 4111–4121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Marco, A.; Heuser, A.M.; Glass, B.; Krausslich, H.G.; Muller, B.; Briggs, J.A. Role of the SP2 domain and its proteolytic cleavage in HIV-1 structural maturation and infectivity. J. Virol. 2012, 86, 13708–13716. [Google Scholar] [CrossRef] [Green Version]
- Wiegers, K.; Rutter, G.; Kottler, H.; Tessmer, U.; Hohenberg, H.; Krausslich, H.G. Sequential steps in human immunodeficiency virus particle maturation revealed by alterations of individual Gag polyprotein cleavage sites. J. Virol. 1998, 72, 2846–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, I.; Hohenberg, H.; Wilk, T.; Wiegers, K.; Grattinger, M.; Muller, B.; Fuller, S.; Krausslich, H.G. A conformational switch controlling HIV-1 morphogenesis. EMBO J. 2000, 19, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharat, T.A.; Menendez, L.R.C.; Hagen, W.J.; Lux, V.; Igonet, S.; Schorb, M.; Schur, F.K.; Krausslich, H.G.; Briggs, J.A. Cryo-electron microscopy of tubular arrays of HIV-1 Gag resolves structures essential for immature virus assembly. Proc. Natl. Acad. Sci. USA 2014, 111, 8233–8238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Lu, W.; Li, F. Pharmacological intervention of HIV-1 maturation. Acta Pharm. Sin. B 2015, 5, 493–499. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Goila-Gaur, R.; Salzwedel, K.; Kilgore, N.R.; Reddick, M.; Matallana, C.; Castillo, A.; Zoumplis, D.; Martin, D.E.; Orenstein, J.M.; et al. PA-457: A potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc. Natl. Acad. Sci. USA 2003, 100, 13555–13560. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Chen, C.H.; Aiken, C. Human immunodeficiency virus type 1 resistance to the small molecule maturation inhibitor 3-O-(3′,3′-dimethylsuccinyl)-betulinic acid is conferred by a variety of single amino acid substitutions at the CA-SP1 cleavage site in Gag. J. Virol. 2006, 80, 12095–12101. [Google Scholar] [CrossRef] [Green Version]
- Purdy, M.D.; Shi, D.; Chrustowicz, J.; Hattne, J.; Gonen, T.; Yeager, M. MicroED structures of HIV-1 Gag CTD-SP1 reveal binding interactions with the maturation inhibitor bevirimat. Proc. Natl. Acad. Sci. USA 2018, 115, 13258–13263. [Google Scholar] [CrossRef] [Green Version]
- Adamson, C.S.; Sakalian, M.; Salzwedel, K.; Freed, E.O. Polymorphisms in Gag spacer peptide 1 confer varying levels of resistance to the HIV- 1 maturation inhibitor bevirimat. Retrovirology 2010, 7, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Baelen, K.; Salzwedel, K.; Rondelez, E.; van Eygen, V.; de Vos, S.; Verheyen, A.; Steegen, K.; Verlinden, Y.; Allaway, G.P.; Stuyver, L.J. Susceptibility of human immunodeficiency virus type 1 to the maturation inhibitor bevirimat is modulated by baseline polymorphisms in Gag spacer peptide 1. Antimicrob. Agents Chemother. 2009, 53, 2185–2188. [Google Scholar] [CrossRef] [Green Version]
- Blair, W.S.; Cao, J.; Fok-Seang, J.; Griffin, P.; Isaacson, J.; Jackson, R.L.; Murray, E.; Patick, A.K.; Peng, Q.; Perros, M.; et al. New small-molecule inhibitor class targeting human immunodeficiency virus type 1 virion maturation. Antimicrob. Agents Chemother. 2009, 53, 5080–5087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waki, K.; Durell, S.R.; Soheilian, F.; Nagashima, K.; Butler, S.L.; Freed, E.O. Structural and functional insights into the HIV-1 maturation inhibitor binding pocket. PLoS Pathog. 2012, 8, e1002997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowicka-Sans, B.; Protack, T.; Lin, Z.; Li, Z.; Zhang, S.; Sun, Y.; Samanta, H.; Terry, B.; Liu, Z.; Chen, Y.; et al. Identification and Characterization of BMS-955176, a Second-Generation HIV-1 Maturation Inhibitor with Improved Potency, Antiviral Spectrum, and Gag Polymorphic Coverage. Antimicrob. Agents Chemother. 2016, 60, 3956–3969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urano, E.; Timilsina, U.; Kaplan, J.A.; Ablan, S.; Ghimire, D.; Pham, P.; Kuruppu, N.; Mandt, R.; Durell, S.R.; Nitz, T.J.; et al. Resistance to Second-Generation HIV-1 Maturation Inhibitors. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Swidorski, J.J.; Nowicka-Sans, B.; Terry, B.; Protack, T.; Lin, Z.; Samanta, H.; Zhang, S.; Li, Z.; Parker, D.D.; et al. C-3 benzoic acid derivatives of C-3 deoxybetulinic acid and deoxybetulin as HIV-1 maturation inhibitors. Bioorg. Med. Chem. 2016, 24, 1757–1770. [Google Scholar] [CrossRef]
- Qian, K.; Bori, I.D.; Chen, C.H.; Huang, L.; Lee, K.H. Anti-AIDS agents 90. novel C-28 modified bevirimat analogues as potent HIV maturation inhibitors. J. Med. Chem. 2012, 55, 8128–8136. [Google Scholar]
- Qian, K.; Kuo, R.Y.; Chen, C.H.; Huang, L.; Morris-Natschke, S.L.; Lee, K.H. Anti-AIDS agents 81. Design, synthesis, and structure-activity relationship study of betulinic acid and moronic acid derivatives as potent HIV maturation inhibitors. J. Med. Chem. 2010, 53, 3133–3141. [Google Scholar]
- Swidorski, J.J.; Liu, Z.; Sit, S.Y.; Chen, J.; Chen, Y.; Sin, N.; Venables, B.L.; Parker, D.D.; Nowicka-Sans, B.; Terry, B.J.; et al. Inhibitors of HIV-1 maturation: Development of structure-activity relationship for C-28 amides based on C-3 benzoic acid-modified triterpenoids. Bioorg. Med. Chem. Lett. 2016, 26, 1925–1930. [Google Scholar] [CrossRef]
- Tang, J.; Jones, S.A.; Jeffery, J.L.; Miranda, S.R.; Galardi, C.M.; Irlbeck, D.M.; Brown, K.W.; McDanal, C.B.; Han, N.; Gao, D.; et al. Synthesis and Biological Evaluation of Macrocyclized Betulin Derivatives as a Novel Class of Anti-HIV-1 Maturation Inhibitors. Open Med. Chem. J. 2014, 8, 23–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urano, E.; Ablan, S.D.; Mandt, R.; Pauly, G.T.; Sigano, D.M.; Schneider, J.P.; Martin, D.E.; Nitz, T.J.; Wild, C.T.; Freed, E.O. Alkyl Amine Bevirimat Derivatives Are Potent and Broadly Active HIV-1 Maturation Inhibitors. Antimicrob. Agents Chemother. 2016, 60, 190–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales-Ramirez, J.; Bogner, J.R.; Molina, J.M.; Lombaard, J.; Dicker, I.B.; Stock, D.A.; DeGrosky, M.; Gartland, M.; Dumitrescu, T.P.; Min, S.; et al. Safety, efficacy, and dose response of the maturation inhibitor GSK3532795 (formerly known as BMS-955176) plus tenofovir/emtricitabine once daily in treatment-naive HIV-1-infected adults: Week 24 primary analysis from a randomized Phase IIb trial. PLoS ONE 2018, 13, e0205368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganser-Pornillos, B.K.; Yeager, M.; Sundquist, W.I. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 2008, 18, 203–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouvenet, N.; Neil, S.J.; Bess, C.; Johnson, M.C.; Virgen, C.A.; Simon, S.M.; Bieniasz, P.D. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006, 4, e435. [Google Scholar] [CrossRef] [Green Version]
- Vlach, J.; Saad, J.S. Trio engagement via plasma membrane phospholipids and the myristoyl moiety governs HIV-1 matrix binding to bilayers. Proc. Natl. Acad. Sci. USA 2013, 110, 3525–3530. [Google Scholar] [CrossRef] [Green Version]
- Chukkapalli, V.; Oh, S.J.; Ono, A. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc. Natl. Acad. Sci. USA 2010, 107, 1600–1605. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.; Garg, H.; Nagashima, K.; Bonifacino, J.S.; Freed, E.O. GGA and Arf proteins modulate retrovirus assembly and release. Mol. Cell 2008, 30, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Saad, J.S.; Loeliger, E.; Luncsford, P.; Liriano, M.; Tai, J.; Kim, A.; Miller, J.; Joshi, A.; Freed, E.O.; Summers, M.F. Point mutations in the HIV-1 matrix protein turn off the myristyl switch. J. Mol. Biol. 2007, 366, 574–585. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Loeliger, E.; Luncsford, P.; Kinde, I.; Beckett, D.; Summers, M.F. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc. Natl. Acad. Sci. USA 2004, 101, 517–522. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.P.; Worthylake, D.; Bancroft, D.P.; Christensen, A.M.; Sundquist, W.I. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: Implications for membrane association and assembly. Proc. Natl. Acad. Sci. USA 1996, 93, 3099–3104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, Z.; Belyaev, A.S.; Fry, E.; Roy, P.; Jones, I.M.; Stuart, D.I. Crystal structure of SIV matrix antigen and implications for virus assembly. Nature 1995, 378, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Tedbury, P.R.; Ablan, S.D.; Freed, E.O. Global rescue of defects in HIV-1 envelope glycoprotein incorporation: Implications for matrix structure. PLoS Pathog. 2013, 9, e1003739. [Google Scholar] [CrossRef] [PubMed]
- Tedbury, P.R.; Novikova, M.; Ablan, S.D.; Freed, E.O. Biochemical evidence of a role for matrix trimerization in HIV-1 envelope glycoprotein incorporation. Proc. Natl. Acad. Sci. USA 2016, 113, E182–E190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfadhli, A.; Barklis, R.L.; Barklis, E. HIV-1 matrix organizes as a hexamer of trimers on membranes containing phosphatidylinositol-(4,5)-bisphosphate. Virology 2009, 387, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol. 2011, 410, 582–608. [Google Scholar] [CrossRef] [Green Version]
- Tedbury, P.R.; Freed, E.O. The role of matrix in HIV-1 envelope glycoprotein incorporation. Trends Microbiol. 2014, 22, 372–378. [Google Scholar] [CrossRef] [Green Version]
- Chojnacki, J.; Waithe, D.; Carravilla, P.; Huarte, N.; Galiani, S.; Enderlein, J.; Eggeling, C. Envelope glycoprotein mobility on HIV-1 particles depends on the virus maturation state. Nat. Commun. 2017, 8, 545. [Google Scholar] [CrossRef] [Green Version]
- Muranyi, W.; Malkusch, S.; Muller, B.; Heilemann, M.; Krausslich, H.G. Super-resolution microscopy reveals specific recruitment of HIV-1 envelope proteins to viral assembly sites dependent on the envelope C-terminal tail. PLoS Pathog. 2013, 9, e1003198. [Google Scholar] [CrossRef]
- Murakami, T.; Freed, E.O. The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc. Natl. Acad. Sci. USA 2000, 97, 343–348. [Google Scholar] [CrossRef] [Green Version]
- Akari, H.; Fukumori, T.; Adachi, A. Cell-dependent requirement of human immunodeficiency virus type 1 gp41 cytoplasmic tail for Env incorporation into virions. J. Virol. 2000, 74, 4891–4893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammano, F.; Kondo, E.; Sodroski, J.; Bukovsky, A.; Gottlinger, H.G. Rescue of human immunodeficiency virus type 1 matrix protein mutants by envelope glycoproteins with short cytoplasmic domains. J. Virol. 1995, 69, 3824–3830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfadhli, A.; Staubus, A.O.; Tedbury, P.R.; Novikova, M.; Freed, E.O.; Barklis, E. Analysis of HIV-1 matrix-envelope cytoplasmic tail interactions. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Buttler, C.A.; Pezeshkian, N.; Fernandez, M.V.; Aaron, J.; Norman, S.; Freed, E.O.; van Engelenburg, S.B. Single molecule fate of HIV-1 envelope reveals late-stage viral lattice incorporation. Nat. Commun. 2018, 9, 1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Chertova, E.; Bess, J., Jr.; Lifson, J.D.; Arthur, L.O.; Liu, J.; Taylor, K.A.; Roux, K.H. Electron tomography analysis of envelope glycoprotein trimers on HIV and simian immunodeficiency virus virions. Proc. Natl. Acad. Sci. USA 2003, 100, 15812–15817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, A.; Ablan, S.D.; Lockett, S.J.; Nagashima, K.; Freed, E.O. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. USA 2004, 101, 14889–14894. [Google Scholar] [CrossRef] [Green Version]
- Chukkapalli, V.; Hogue, I.B.; Boyko, V.; Hu, W.S.; Ono, A. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient gag membrane binding. J. Virol. 2008, 82, 2405–2417. [Google Scholar] [CrossRef] [Green Version]
- Haffar, O.; Dubrovsky, L.; Lowe, R.; Berro, R.; Kashanchi, F.; Godden, J.; Vanpouille, C.; Bajorath, J.; Bukrinsky, M. Oxadiazols: A new class of rationally designed anti-human immunodeficiency virus compounds targeting the nuclear localization signal of the viral matrix protein. J. Virol. 2005, 79, 13028–13036. [Google Scholar] [CrossRef] [Green Version]
- Alfadhli, A.; McNett, H.; Eccles, J.; Tsagli, S.; Noviello, C.; Sloan, R.; Lopez, C.S.; Peyton, D.H.; Barklis, E. Analysis of small molecule ligands targeting the HIV-1 matrix protein-RNA binding site. J. Biol. Chem. 2013, 288, 666–676. [Google Scholar] [CrossRef] [Green Version]
- Zentner, I.; Sierra, L.J.; Fraser, A.K.; Maciunas, L.; Mankowski, M.K.; Vinnik, A.; Fedichev, P.; Ptak, R.G.; Martin-Garcia, J.; Cocklin, S. Identification of a small-molecule inhibitor of HIV-1 assembly that targets the phosphatidylinositol (4,5)-bisphosphate binding site of the HIV-1 matrix protein. ChemMedChem 2013, 8, 426–432. [Google Scholar] [CrossRef]
- Zentner, I.; Sierra, L.J.; Maciunas, L.; Vinnik, A.; Fedichev, P.; Mankowski, M.K.; Ptak, R.G.; Martin-Garcia, J.; Cocklin, S. Discovery of a small-molecule antiviral targeting the HIV-1 matrix protein. Bioorg. Med. Chem. Lett. 2013, 23, 1132–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedbury, P.R.; Novikova, M.; Alfadhli, A.; Hikichi, Y.; Kagiampakis, I.; KewalRamani, V.N.; Barklis, E.; Freed, E.O. HIV-1 Matrix Trimerization-Impaired Mutants Are Rescued by Matrix Substitutions That Enhance Envelope Glycoprotein Incorporation. J. Virol. 2019, 94. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.E.; Samal, A.B.; Vlach, J.; Saad, J.S. Solution Structure and Membrane Interaction of the Cytoplasmic Tail of HIV-1 gp41 Protein. Structure 2017, 25, 1708–1718 e1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rihn, S.J.; Wilson, S.J.; Loman, N.J.; Alim, M.; Bakker, S.E.; Bhella, D.; Gifford, R.J.; Rixon, F.J.; Bieniasz, P.D. Extreme genetic fragility of the HIV-1 capsid. PLoS Pathog. 2013, 9, e1003461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momany, C.; Kovari, L.C.; Prongay, A.J.; Keller, W.; Gitti, R.K.; Lee, B.M.; Gorbalenya, A.E.; Tong, L.; McClure, J.; Ehrlich, L.S.; et al. Crystal structure of dimeric HIV-1 capsid protein. Nat. Struct. Biol. 1996, 3, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Gitti, R.K.; Lee, B.M.; Walker, J.; Summers, M.F.; Yoo, S.; Sundquist, W.I. Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science 1996, 273, 231–235. [Google Scholar] [CrossRef]
- Luban, J.; Bossolt, K.L.; Franke, E.K.; Kalpana, G.V.; Goff, S.P. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 1993, 73, 1067–1078. [Google Scholar] [CrossRef]
- Briggs, J.A.; Krausslich, H.G. The molecular architecture of HIV. J. Mol. Biol. 2011, 410, 491–500. [Google Scholar] [CrossRef]
- Briggs, J.A.; Riches, J.D.; Glass, B.; Bartonova, V.; Zanetti, G.; Krausslich, H.G. Structure and assembly of immature HIV. Proc. Natl. Acad. Sci. USA 2009, 106, 11090–11095. [Google Scholar] [CrossRef] [Green Version]
- Mattei, S.; Glass, B.; Hagen, W.J.; Krausslich, H.G.; Briggs, J.A. The structure and flexibility of conical HIV-1 capsids determined within intact virions. Science 2016, 354, 1434–1437. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Cheng, A.; Yeager, M. Structure of full-length HIV-1 CA: A model for the mature capsid lattice. Cell 2007, 131, 70–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forshey, B.M.; von Schwedler, U.; Sundquist, W.I.; Aiken, C. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 2002, 76, 5667–5677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arhel, N.J.; Souquere-Besse, S.; Munier, S.; Souque, P.; Guadagnini, S.; Rutherford, S.; Prevost, M.C.; Allen, T.D.; Charneau, P. HIV-1 DNA Flap formation promotes uncoating of the pre-integration complex at the nuclear pore. EMBO J. 2007, 26, 3025–3037. [Google Scholar] [CrossRef] [PubMed]
- Rawle, D.J.; Harrich, D. Toward the “unravelling” of HIV: Host cell factors involved in HIV-1 core uncoating. PLoS Pathog. 2018, 14, e1007270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.T.; Summers, B.J.; Xu, C.; Perilla, J.R.; Malikov, V.; Naghavi, M.H.; Xiong, Y. FEZ1 Is Recruited to a Conserved Cofactor Site on Capsid to Promote HIV-1 Trafficking. Cell Rep. 2019, 28, 2373–2385.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malikov, V.; da Silva, E.S.; Jovasevic, V.; Bennett, G.; Vieira, D.A.D.A.; Schulte, B.; Diaz-Griffero, F.; Walsh, D.; Naghavi, M.H. HIV-1 capsids bind and exploit the kinesin-1 adaptor FEZ1 for inward movement to the nucleus. Nat. Commun. 2015, 6, 6660. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.; Machado, A.K.; Lyonnais, S.; Chamontin, C.; Gartner, K.; Leger, T.; Henriquet, C.; Garcia, C.; Portilho, D.M.; Pugniere, M.; et al. Transportin-1 binds to the HIV-1 capsid via a nuclear localization signal and triggers uncoating. Nat. Microbiol. 2019, 4, 1840–1850. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Moncorge, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hue, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef] [Green Version]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef]
- Sayah, D.M.; Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 2004, 430, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Wu, J.; Wu, Y.T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Loeliger, E.; Kinde, I.; Kyere, S.; Mayo, K.; Barklis, E.; Sun, Y.; Huang, M.; Summers, M.F. Antiviral inhibition of the HIV-1 capsid protein. J. Mol. Biol. 2003, 327, 1013–1020. [Google Scholar] [CrossRef]
- Kelly, B.N.; Kyere, S.; Kinde, I.; Tang, C.; Howard, B.R.; Robinson, H.; Sundquist, W.I.; Summers, M.F.; Hill, C.P. Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J. Mol. Biol. 2007, 373, 355–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sticht, J.; Humbert, M.; Findlow, S.; Bodem, J.; Muller, B.; Dietrich, U.; Werner, J.; Krausslich, H.G. A peptide inhibitor of HIV-1 assembly in vitro. Nat. Struct. Mol. Biol. 2005, 12, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, Q.; Bhattacharya, S.; Waheed, A.A.; Tong, X.; Hong, A.; Heck, S.; Curreli, F.; Goger, M.; Cowburn, D.; et al. A cell-penetrating helical peptide as a potential HIV-1 inhibitor. J. Mol. Biol. 2008, 378, 565–580. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Zhang, H.; Debnath, A.K.; Cowburn, D. Solution structure of a hydrocarbon stapled peptide inhibitor in complex with monomeric C-terminal domain of HIV-1 capsid. J. Biol. Chem. 2008, 283, 16274–16278. [Google Scholar] [CrossRef] [Green Version]
- Fader, L.D.; Bethell, R.; Bonneau, P.; Bos, M.; Bousquet, Y.; Cordingley, M.G.; Coulombe, R.; Deroy, P.; Faucher, A.M.; Gagnon, A.; et al. Discovery of a 1,5-dihydrobenzo[b][1,4]diazepine-2,4-dione series of inhibitors of HIV-1 capsid assembly. Bioorg. Med. Chem. Lett. 2011, 21, 398–404. [Google Scholar] [CrossRef]
- Lemke, C.T.; Titolo, S.; von Schwedler, U.; Goudreau, N.; Mercier, J.F.; Wardrop, E.; Faucher, A.M.; Coulombe, R.; Banik, S.S.; Fader, L.; et al. Distinct effects of two HIV-1 capsid assembly inhibitor families that bind the same site within the N-terminal domain of the viral CA protein. J. Virol. 2012, 86, 6643–6655. [Google Scholar] [CrossRef] [Green Version]
- Blair, W.S.; Pickford, C.; Irving, S.L.; Brown, D.G.; Anderson, M.; Bazin, R.; Cao, J.; Ciaramella, G.; Isaacson, J.; Jackson, L.; et al. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 2010, 6, e1001220. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhou, J.; Shah, V.B.; Aiken, C.; Whitby, K. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J. Virol. 2011, 85, 542–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, A.; Alam, S.L.; Fricke, T.; Zadrozny, K.; Sedzicki, J.; Taylor, A.B.; Demeler, B.; Pornillos, O.; Ganser-Pornillos, B.K.; Diaz-Griffero, F.; et al. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc. Natl. Acad. Sci. USA 2014, 111, 18625–18630. [Google Scholar] [CrossRef] [Green Version]
- Price, A.J.; Jacques, D.A.; McEwan, W.A.; Fletcher, A.J.; Essig, S.; Chin, J.W.; Halambage, U.D.; Aiken, C.; James, L.C. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014, 10, e1004459. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Engelman, A.N. Capsid-Dependent Host Factors in HIV-1 Infection. Trends Microbiol. 2017, 25, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, S.; Ramalho, R.; Aiken, C.; Rousso, I. PF74 Reinforces the HIV-1 Capsid To Impair Reverse Transcription-Induced Uncoating. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.P.; Francis, A.C.; Meuser, M.E.; Mankowski, M.; Ptak, R.G.; Rashad, A.A.; Melikyan, G.B.; Cocklin, S. Exploring Modifications of an HIV-1 Capsid Inhibitor: Design, Synthesis, and Mechanism of Action. J. Drug Des. Res. 2018, 5, 1070. [Google Scholar]
- Xu, J.P.; Branson, J.D.; Lawrence, R.; Cocklin, S. Identification of a small molecule HIV-1 inhibitor that targets the capsid hexamer. Bioorg. Med. Chem. Lett. 2016, 26, 824–828. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Zalloum, W.A.; Meuser, M.E.; Jing, L.; Kang, D.; Chen, C.H.; Tian, Y.; Zhang, F.; Cocklin, S.; Lee, K.H.; et al. Discovery of phenylalanine derivatives as potent HIV-1 capsid inhibitors from click chemistry-based compound library. Eur. J. Med. Chem. 2018, 158, 478–492. [Google Scholar] [CrossRef]
- Jiang, X.; Wu, G.; Zalloum, W.A.; Meuser, M.E.; Dick, A.; Sun, L.; Chen, C.H.; Kang, D.; Jing, L.; Jia, R.; et al. Discovery of novel 1,4-disubstituted 1,2,3-triazole phenylalanine derivatives as HIV-1 capsid inhibitors. RSC Adv. 2019, 9, 28961–28986. [Google Scholar] [CrossRef] [Green Version]
- Lamorte, L.; Titolo, S.; Lemke, C.T.; Goudreau, N.; Mercier, J.F.; Wardrop, E.; Shah, V.B.; von Schwedler, U.K.; Langelier, C.; Banik, S.S.; et al. Discovery of novel small-molecule HIV-1 replication inhibitors that stabilize capsid complexes. Antimicrob. Agents Chemother. 2013, 57, 4622–4631. [Google Scholar] [CrossRef] [Green Version]
- Fricke, T.; Buffone, C.; Opp, S.; Valle-Casuso, J.; Diaz-Griffero, F. BI-2 destabilizes HIV-1 cores during infection and Prevents Binding of CPSF6 to the HIV-1 Capsid. Retrovirology 2014, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- Lemke, C.T.; Titolo, S.; Goudreau, N.; Faucher, A.M.; Mason, S.W.; Bonneau, P. A novel inhibitor-binding site on the HIV-1 capsid N-terminal domain leads to improved crystallization via compound-mediated dimerization. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, J.; Halambage, U.D.; Jurado, K.A.; Jamin, A.V.; Wang, Y.; Engelman, A.N.; Aiken, C. Inhibition of HIV-1 Maturation via Small-Molecule Targeting of the Amino-Terminal Domain in the Viral Capsid Protein. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thenin-Houssier, S.; de Vera, I.M.; Pedro-Rosa, L.; Brady, A.; Richard, A.; Konnick, B.; Opp, S.; Buffone, C.; Fuhrmann, J.; Kota, S.; et al. Ebselen, a Small-Molecule Capsid Inhibitor of HIV-1 Replication. Antimicrob. Agents Chemother. 2016, 60, 2195–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, W.C.; Link, J.O.; Mulato, A.; Niedziela-Majka, A.; Rowe, W.; Somoza, J.R.; Villasenor, A.G.; Yant, S.R.; Zhang, J.R.; Zheng, J. Discovery of Novel Potent HIV Capsid Inhibitors with Long-acting Potential. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, Seattle, WA, USA, 13–16 February 2017. [Google Scholar]
- Yant, S.R.; Mulato, A.; Hansen, D.; Tse, W.C.; Niedziela-Majka, A.; Zhang, J.R.; Stepan, G.J.; Jin, D.; Wong, M.H.; Perreira, J.M.; et al. A highly potent long-acting small-molecule HIV-1 capsid inhibitor with efficacy in a humanized mouse model. Nat. Med. 2019, 25, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Yant, S.R.; Mulato, A.; Stepan, G.; Villasenor, A.G.; Jin, D.; Margot, N.A. GS-6207, a potent and selective first-in-class long-acting HIV-1 capsid inhibitor. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, Seattle, WA, USA, 4–7 March 2019. [Google Scholar]
- Singh, K.; Gallazzi, F.; Hill, K.J.; Burke, D.H.; Lange, M.J.; Quinn, T.P.; Neogi, U.; Sonnerborg, A. GS-CA Compounds: First-In-Class HIV-1 Capsid Inhibitors Covering Multiple Grounds. Front. Microbiol. 2019, 10, 1227. [Google Scholar] [CrossRef] [PubMed]
- Bell, N.M.; Lever, A.M. HIV Gag polyprotein: Processing and early viral particle assembly. Trends Microbiol. 2013, 21, 136–144. [Google Scholar] [CrossRef]
- Darlix, J.L.; Godet, J.; Ivanyi-Nagy, R.; Fosse, P.; Mauffret, O.; Mely, Y. Flexible nature and specific functions of the HIV-1 nucleocapsid protein. J. Mol. Biol. 2011, 410, 565–581. [Google Scholar] [CrossRef]
- Levin, J.G.; Guo, J.; Rouzina, I.; Musier-Forsyth, K. Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: Critical role in reverse transcription and molecular mechanism. Prog. Nucleic Acid Res. Mol. Biol. 2005, 80, 217–286. [Google Scholar]
- Levin, J.G.; Mitra, M.; Mascarenhas, A.; Musier-Forsyth, K. Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription. RNA Biol. 2010, 7, 754–774. [Google Scholar] [CrossRef] [Green Version]
- Dorfman, T.; Luban, J.; Goff, S.P.; Haseltine, W.A.; Gottlinger, H.G. Mapping of functionally important residues of a cysteine-histidine box in the human immunodeficiency virus type 1 nucleocapsid protein. J. Virol. 1993, 67, 6159–6169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorelick, R.J.; Nigida, S.M., Jr.; Bess, J.W., Jr.; Arthur, L.O.; Henderson, L.E.; Rein, A. Noninfectious human immunodeficiency virus type 1 mutants deficient in genomic RNA. J. Virol. 1990, 64, 3207–3211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Rocquigny, H.; Shvadchak, V.; Avilov, S.; Dong, C.Z.; Dietrich, U.; Darlix, J.L.; Mely, Y. Targeting the viral nucleocapsid protein in anti-HIV-1 therapy. Mini Rev. Med. Chem. 2008, 8, 24–35. [Google Scholar] [PubMed]
- Rice, W.G.; Schaeffer, C.A.; Harten, B.; Villinger, F.; South, T.L.; Summers, M.F.; Henderson, L.E.; Bess, J.W., Jr.; Arthur, L.O.; McDougal, J.S.; et al. Inhibition of HIV-1 infectivity by zinc-ejecting aromatic C-nitroso compounds. Nature 1993, 361, 473–475. [Google Scholar] [CrossRef]
- Rice, W.G.; Supko, J.G.; Malspeis, L.; Buckheit, R.W., Jr.; Clanton, D.; Bu, M.; Graham, L.; Schaeffer, C.A.; Turpin, J.A.; Domagala, J.; et al. Inhibitors of HIV nucleocapsid protein zinc fingers as candidates for the treatment of AIDS. Science 1995, 270, 1194–1197. [Google Scholar] [CrossRef]
- Rice, W.G.; Turpin, J.A. Virus-encoded Zinc Fingers as Targets for Antiviral Chemotherapy. Rev. Med. Virol. 1996, 6, 187–199. [Google Scholar] [CrossRef]
- Turpin, J.A.; Song, Y.; Inman, J.K.; Huang, M.; Wallqvist, A.; Maynard, A.; Covell, D.G.; Rice, W.G.; Appella, E. Synthesis and biological properties of novel pyridinioalkanoyl thiolesters (PATE) as anti-HIV-1 agents that target the viral nucleocapsid protein zinc fingers. J. Med. Chem. 1999, 42, 67–86. [Google Scholar] [CrossRef]
- Jenkins, L.M.; Byrd, J.C.; Hara, T.; Srivastava, P.; Mazur, S.J.; Stahl, S.J.; Inman, J.K.; Appella, E.; Omichinski, J.G.; Legault, P. Studies on the mechanism of inactivation of the HIV-1 nucleocapsid protein NCp7 with 2-mercaptobenzamide thioesters. J. Med. Chem. 2005, 48, 2847–2858. [Google Scholar] [CrossRef]
- Quintal, S.M.; dePaula, Q.A.; Farrell, N.P. Zinc finger proteins as templates for metal ion exchange and ligand reactivity. Chemical and biological consequences. Metallomics 2011, 3, 121–139. [Google Scholar] [CrossRef]
- Pannecouque, C.; Szafarowicz, B.; Volkova, N.; Bakulev, V.; Dehaen, W.; Mely, Y.; Daelemans, D. Inhibition of HIV-1 replication by a bis-thiadiazolbenzene-1,2-diamine that chelates zinc ions from retroviral nucleocapsid zinc fingers. Antimicrob. Agents Chemother. 2010, 54, 1461–1468. [Google Scholar] [CrossRef] [Green Version]
- Vercruysse, T.; Basta, B.; Dehaen, W.; Humbert, N.; Balzarini, J.; Debaene, F.; Sanglier-Cianferani, S.; Pannecouque, C.; Mely, Y.; Daelemans, D. A phenyl-thiadiazolylidene-amine derivative ejects zinc from retroviral nucleocapsid zinc fingers and inactivates HIV virions. Retrovirology 2012, 9, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, J.A.; Holler, T.P.; Sanchez, J.; Gogliotti, R.; Maloney, L.; Reily, M.D. Biophysical characterization of zinc ejection from HIV nucleocapsid protein by anti-HIV 2,2′-dithiobis[benzamides] and benzisothiazolones. J. Med. Chem. 1996, 39, 4313–4320. [Google Scholar] [CrossRef] [PubMed]
- Maynard, A.T.; Covell, D.G. Reactivity of zinc finger cores: Analysis of protein packing and electrostatic screening. J. Am. Chem. Soc. 2001, 123, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Anzellotti, A.I.; Liu, Q.; Bloemink, M.J.; Scarsdale, J.N.; Farrell, N. Targeting retroviral Zn finger-DNA interactions: A small-molecule approach using the electrophilic nature of trans-platinum-nucleobase compounds. Chem. Biol. 2006, 13, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, M.; Kovalenko, L.; Lyonnais, S.; Antaki, D.; Torbett, B.E.; Botta, M.; Mirambeau, G.; Mely, Y. Nucleocapsid Protein: A Desirable Target for Future Therapies Against HIV-1. Curr. Top. Microbiol. Immunol. 2015, 389, 53–92. [Google Scholar]
- Breuer, S.; Chang, M.W.; Yuan, J.; Torbett, B.E. Identification of HIV-1 inhibitors targeting the nucleocapsid protein. J. Med. Chem. 2012, 55, 4968–4977. [Google Scholar] [CrossRef] [Green Version]
- Goudreau, N.; Hucke, O.; Faucher, A.M.; Grand-Maitre, C.; Lepage, O.; Bonneau, P.R.; Mason, S.W.; Titolo, S. Discovery and structural characterization of a new inhibitor series of HIV-1 nucleocapsid function: NMR solution structure determination of a ternary complex involving a 2:1 inhibitor/NC stoichiometry. J. Mol. Biol. 2013, 425, 1982–1998. [Google Scholar] [CrossRef]
- Mori, M.; Nucci, A.; Lang, M.C.; Humbert, N.; Boudier, C.; Debaene, F.; Sanglier-Cianferani, S.; Catala, M.; Schult-Dietrich, P.; Dietrich, U.; et al. Functional and structural characterization of 2-amino-4-phenylthiazole inhibitors of the HIV-1 nucleocapsid protein with antiviral activity. ACS Chem. Biol. 2014, 9, 1950–1955. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, S.H.; Park, J.A.; Yu, K.L.; Jang, S.I.; Kim, B.S.; Lee, E.S.; You, J.C. Identification and characterization of a new type of inhibitor against the human immunodeficiency virus type-1 nucleocapsid protein. Retrovirology 2015, 12, 90. [Google Scholar] [CrossRef]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Cote, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- VerPlank, L.; Bouamr, F.; LaGrassa, T.J.; Agresta, B.; Kikonyogo, A.; Leis, J.; Carter, C.A. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc. Natl. Acad. Sci. USA 2001, 98, 7724–7729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, A.N.; Waheed, A.A.; Ablan, S.D.; Huang, W.; Newton, A.; Petropoulos, C.J.; Brindeiro, R.D.; Freed, E.O. Elucidation of the Molecular Mechanism Driving Duplication of the HIV-1 PTAP Late Domain. J. Virol. 2016, 90, 768–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Liu, X.; Li, Z.; Zhan, P.; de Clercq, E. TSG101: A novel anti-HIV-1 drug target. Curr. Med. Chem. 2010, 17, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Liu, F.; Im, Y.J.; Stephen, A.G.; Fivash, M.J.; Waheed, A.A.; Freed, E.O.; Fisher, R.J.; Hurley, J.H.; Burke, T.R., Jr. Elucidation of New Binding Interactions with the Tumor Susceptibility Gene 101 (Tsg101) Protein Using Modified HIV-1 Gag-p6 Derived Peptide Ligands. ACS Med. Chem. Lett. 2011, 2, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Munshi, U.M.; Kim, J.; Nagashima, K.; Hurley, J.H.; Freed, E.O. An Alix fragment potently inhibits HIV-1 budding: Characterization of binding to retroviral YPXL late domains. J. Biol. Chem. 2007, 282, 3847–3855. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Stephen, A.G.; Adamson, C.S.; Gousset, K.; Aman, M.J.; Freed, E.O.; Fisher, R.J.; Burke, T.R., Jr. Hydrazone- and hydrazide-containing N-substituted glycines as peptoid surrogates for expedited library synthesis: Application to the preparation of Tsg101-directed HIV-1 budding antagonists. Org. Lett. 2006, 8, 5165–5168. [Google Scholar] [CrossRef] [Green Version]
- Tavassoli, A.; Lu, Q.; Gam, J.; Pan, H.; Benkovic, S.J.; Cohen, S.N. Inhibition of HIV budding by a genetically selected cyclic peptide targeting the Gag-TSG101 interaction. ACS Chem. Biol. 2008, 3, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Strickland, M.; Ehrlich, L.S.; Watanabe, S.; Khan, M.; Strub, M.P.; Luan, C.H.; Powell, M.D.; Leis, J.; Tjandra, N.; Carter, C.A. Tsg101 chaperone function revealed by HIV-1 assembly inhibitors. Nat. Commun. 2017, 8, 1391. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protease (PR) and Maturation Inhibitors (MI) | Target and Binding Site | Mechanism of Action | Antiviral Potency (IC50) | CC50 | Clinical Status |
|---|---|---|---|---|---|
| Bevirimat (BVM) | CA-SP1 junction site | Stabilizes six-helix bundle in CA hexamer and prevents CA-SP1 cleavage | ~10 nM | ~25 μM | Failed in phase IIb due to resistance mutations in CA-SP1 |
| PF-46396 | CA-SP1 junction site | Implications for Gag assembly, release and virus replication | 0.005–7 μM (PBMCs) | 17 μM (PBMCs) | Not entered |
| GSK3532795 | CA-SP1 junction site | Late-stage inhibition of CA-SP1 cleavage | 21 nM | 2.3 to > 15 μM | Post phase IIb termination due to high rates of adverse gastrointestinal events, and frequency of treatment-emergent nucleoside reverse transcriptase inhibitor (NRTI) resistance |
| Matrix (MA) Inhibitors | Target and Binding Site | Mechanism of Action | Antiviral Potency (IC50) | CC50 | Clinical Status |
| (Thiadiazolane class) e.g., TD2 | MA RNA binding site | RNA displacement | 1–5 μM | 5-20 μM | Not entered |
| Compound 7 and 14 | MA PI[4,5]P2 binding site | PI[4,5]P2 displacement | 7.5–15.6 µM (group M isolates) | Compound 7 and 14 = >100 µM (PBMCs); compound 7 = >1 mM (293T cells) | Not entered |
| Capsid (CA) Inhibitor | Target and Binding Site | Mechanism of Action | Antiviral Potency (IC50) | CC50 | Clinical Status |
| CAP-1 | NTD | Blocks CA self-association in late events | EC95 ≈ 100 μM | >100 μM | Not entered |
| Peptide Inhibitors (CAI, NYAD-1) | CTD | Blocks assembly of immature and mature-like particles | N.D. (CAI) = 4.29–21.6 μM (NYAD-1 PBMCs) | N.D. (CAI) N.D. (NYAD-1) | Not entered |
| BD-1 | NTD | Blocks CA assembly | 70 ± 30 nM | >28 μM | Not entered |
| BM-1 | NTD | Blocks HIV-1 maturation | 62 ± 23 nM | >20 μM | Not entered |
| PF74 | NTD-CTD | Stabilizes CA core in early-stage and inhibits reverse transcription. Distorts CA lattice in the late stage, causing aberrant virus morphology that does not undergo maturation | 80–640 nM (PBMCs) | >10 μM (PBMCs) | Not entered |
| BI Compounds (BI-1, BI-2) | NTD | Destabilizes HIV-1 capsid by interfering in early and late events | 7.5 ± 2.1 μM (BI-1) 1.4 ± 0.66 μM (BI-2) | >91 μM (BI-1) >76 μM (BI-2) | Not entered |
| C1 | NTD | Inhibits HIV-1 replication in late events by disrupting the assembly of the mature capsid | 57 µM | N.D. | Not entered |
| Ebselen | Undetermined | Reverse transcription inhibition and impaired uncoating | 3.37 µM | >30 μM (PBMCs) | Not entered |
| GS-CA1 and GS-6207 | NTD-CTD | Most likely, stabilizes CA core in early-stage and inhibits reverse transcription. Probably, distorts CA lattice in the late stage, causing aberrant virus morphology that does not undergo maturation | 140 pM (GS-CA1, PBMCs) 100 pM (GS-6207, MT-4 cells) | 27 µM (GS-6207) | Phase 1b (GS-6207) |
| Nucleocapsid (NC) Inhibitors | Target and Binding Site | Mechanism of Action | Antiviral Potency (IC50) | CC50 | Clinical Status |
| NOBA | Zinc finger | Class 1 - electrophilic attack of the zinc fingers | N.D. | 10.6 µM | Not entered |
| DIBA-1 | Zinc finger | Class 1 - electrophilic attack of the zinc fingers | 2.3 µM | >200 µM | Not entered |
| PATE-45 | Zinc finger | Class 1 - electrophilic attack of the zinc fingers | 6.2 µM | >316 µM | Not entered |
| SAMT-19 | Zinc finger | Class 1 - electrophilic attack of the zinc fingers | 2.9 µM | 461 µM | Not entered |
| [SP-4-2]-[PtCl(NH3) (quin)(9-EtGH)] | Zinc finger | Class 3 - covalent binding of Cys residues by platinum | 41.9 µM | >200 µM | Not entered |
| NVO38 | Zinc finger | Class 2 - zinc chelation | 17 µM | >300 µM | Not entered |
| WDO-217 | Zinc finger | Class 1 - electrophilic attack of the zinc fingers | 7.9 µM | 72 µM | Not entered |
| Compound 3 | Two molecules bind each zinc knuckle of the NC | Mimicking the guanosine base found in many reported NC complex structures | 0.95 μM (NC-oligonucleotide binding assay) | N.D. | Not entered |
| A1752 | NC | Inhibits NC-mediated dimerization of Psi RNA and cTAR DNA destabilization. Inhibits also proper Gag processing | ~1 µM | >50 μM | Not entered |
| Late domains (P6) Inhibitors | Target and Binding Site | Mechanism of Action | Antiviral Potency (IC50) | CC50 | Clinical Status |
| Cyclic peptide 11 | P6-Tsg101 interface | Blocking the p6-Tsg101 interaction | 7 µM | N.D. | Not entered |
| N16 | Ubiquitin E2 variant domain of Tsg101 | Reduces Gag assembly and virus production in vitro | EC50 between 25 and 50 μM (p24 ELISA) | >50 μM | Phase I as a proton pump inhibitor |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dick, A.; Cocklin, S. Recent Advances in HIV-1 Gag Inhibitor Design and Development. Molecules 2020, 25, 1687. https://doi.org/10.3390/molecules25071687
Dick A, Cocklin S. Recent Advances in HIV-1 Gag Inhibitor Design and Development. Molecules. 2020; 25(7):1687. https://doi.org/10.3390/molecules25071687
Chicago/Turabian StyleDick, Alexej, and Simon Cocklin. 2020. "Recent Advances in HIV-1 Gag Inhibitor Design and Development" Molecules 25, no. 7: 1687. https://doi.org/10.3390/molecules25071687
APA StyleDick, A., & Cocklin, S. (2020). Recent Advances in HIV-1 Gag Inhibitor Design and Development. Molecules, 25(7), 1687. https://doi.org/10.3390/molecules25071687