
H/D Isotope Effects on 1H-NMR Chemical Shifts in Cyclic Heterodimers and Heterotrimers of Phosphinic and Phosphoric Acids


 and
and
Abstract
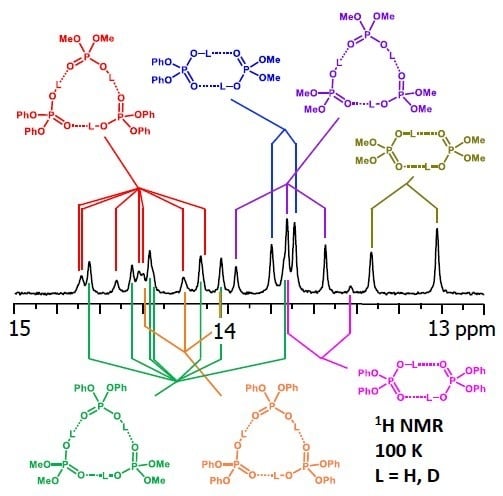
:
1. Introduction
2. Results
2.1. Non-Deuterated Complexes
2.2. Partially Deuterated Complexes
3. Discussion
3.1. Hydrogen Bond Strength
3.2. Cooperativity And Anticooperativity
3.3. A case for the Tetramer
4. Materials and Methods
4.1. Sample Preparation
4.2. NMR Measurements
4.3. NMR Spectra Deconvolution
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shipman, S.T.; Douglass, P.C.; Yoo, H.S.; Hinkle, C.E.; Mierzejewski, E.L.; Pate, B.H. Vibrational dynamics of carboxylic acid dimers in gas and dilute solution. Phys. Chem. Chem. Phys. 2007, 9, 4572–4586. [Google Scholar] [CrossRef] [PubMed]
- Seifert, G.; Patzlaff, T.; Graener, H. Picosecond vibrational energy transfer observed in the CH and OH stretching region of stearic acid dimers in liquid solution. J. Mol. Liquids 2003, 102, 227–240. [Google Scholar] [CrossRef]
- Gaywood, A.; Wood, P.A.; McNab, H. Cambridge Structural Database entry BENZAC20, CCDC 1412517: Experimental Crystal Structure Determination. 2015. Available online: https://search.datacite.org/works/10.5517/CC1JDV2V (accessed on 19 April 2020).
- Steiner, T. Six-fold phenyl embrace in crystalline 3,3,3-triphenylpropionic acid. J. Chem. Cryst. 1999, 29, 1235–1237. [Google Scholar] [CrossRef]
- Allan, D.R.; Clark, S.J. Impeded dimer formation in the high-pressure crystal structure of formic acid. Phys. Rev. Lett. 1999, 82, 3464–3467. [Google Scholar] [CrossRef]
- Nahringbauer, I. Hydrogen bond studies. 39. Reinvestigation of the crystal structure of acetic acid (at +5 degrees C and -190 degrees C). Acta Chem. Scand. 1970, 24, 453–462. [Google Scholar] [CrossRef]
- Fenske, D.; Mattes, R.; Lons, J.; Tebbe, K.F. Die Kristallstruktur von Diphenylphosphinsaure. Chem. Ber. 1973, 106, 1139–1144. [Google Scholar] [CrossRef]
- Ioannou, V. Dimethylphosphinato and dimethylarsinato complexes of Sb(III) and Bi(III) and their chemistry. Monatsh. Chem. 2012, 143, 1349–1356. [Google Scholar] [CrossRef]
- Denisov, G.S.; Tokhadze, K.G. Ultrastrong hydrogen bond in gas phase. Dimer of dimethylphosphinic acid. Dokl. Phys. Chem. 1994, 337, 117–119. [Google Scholar]
- Asfin, R.E.; Denisov, G.S.; Tokhadze, K.G. The infrared spectra and enthalpies of strongly bound dimers of phosphinic acids in the gas phase. (CH2Cl)2POOH and (C6H5)2POOH. J. Mol. Struct. 2002, 608, 161–168. [Google Scholar] [CrossRef]
- Asfin, R.E.; Denisov, G.S.; Poplevchenkov, D.N.; Tokhadze, K.G.; Velikanova, T.V. IR ν(OH) band and dimerization of phosphorous acids in the gas phase and solid state. Pol. J. Chem. 2002, 76, 1223–1231. [Google Scholar]
- Khaikin, L.S.; Grikina, O.E.; Vilkov, L.V.; Golubinskii, A.V.; Atavin, E.G.; Asfin, R.E.; Denisov, G.S. Gas-phase electron diffraction study of cyclic dimer of dimethylphosphinic acid (Me2P(O)OH)2 using quantum-chemical data and a priori force field. J. Mol. Struct. 2003, 658, 153–170. [Google Scholar] [CrossRef]
- Khaikin, L.S.; Grikina, O.E.; Golubinskii, A.V.; Vilkov, L.V.; Atavin, E.G.; Asfin, R.E.; Denisov, G.S. Geometry of a strong hydrogen bond as determined by gas-phase electron diffraction: The cyclic dimer of di-methylphosphinic acid. Dokl. Phys. Chem. 2003, 390, 158–162. [Google Scholar] [CrossRef]
- Detering, C.; Tolstoy, P.M.; Golubev, N.S.; Denisov, G.S.; Limbach, H.H. Vicinal H/D isotope effects in NMR spectra of complexes with coupled hydrogen bonds: Phosphoric acids. Dokl. Phys. Chem. 2001, 379, 191–193. [Google Scholar] [CrossRef]
- Mulloyarova, V.V.; Giba, I.S.; Kostin, M.A.; Denisov, G.S.; Shenderovich, I.G.; Tolstoy, P.M. Cyclic trimers of phosphinic acids in polar aprotic solvent: Symmetry, chirality and H/D isotope effects on NMR chemical shifts. Phys. Chem. Chem. Phys. 2018, 20, 4901–4910. [Google Scholar] [CrossRef]
- Saur, W.; Crespi, H.L.; Katz, J.J. Vicinal deuterium isotope effects on proton chemical shifts. J. Magn. Reson. 1970, 2, 47–49. [Google Scholar] [CrossRef]
- Tolstoy, P.M.; Schah-Mohammedi, P.; Smirnov, S.N.; Golubev, N.S.; Denisov, G.S.; Limbach, H.-H. Characterization of fluxional hydrogen-bonded complexes of acetic acid and acetate by NMR: Geometries and isotope and solvent effects. J. Am. Chem. Soc. 2004, 126, 5621–5634. [Google Scholar] [CrossRef]
- Mulloyarova, V.V.; Giba, I.S.; Denisov, G.S.; Tolstoy, P.M. Conformational mobility and proton transfer in hydrogen-bonded dimers and trimers of phosphinic and phosphoric acids. J. Phys. Chem. A 2019, 123, 6761–6771. [Google Scholar] [CrossRef]
- Klein, O.; Aguilar–Parrilla, F.; Lopez del Amo, J.M.; Jagerovic, N.; Elguero, J.; Limbach, H.H. Dynamic NMR study of the mechanisms of double, triple and quadruple proton and deuteron transfer in cyclic hydrogen bonded solids of pyrazole derivatives. J. Am. Chem. Soc. 2004, 126, 11718–11732. [Google Scholar] [CrossRef] [Green Version]
- Limbach, H.H.; Seiffert, W. Dynamic processes in systems with hydrogen bonds. I. 1H–NMR spectroscopic study of the cis–trans equilibrium and the hydrogen bond association of N,N’-bis(pentadeuterophenyl)-1-amino-3-iminopropene in carbon disulfide. Ber. Bunsen Ges. Phys. Chem. 1974, 78, 532–537. [Google Scholar]
- Forsting, T.; Zischang, J.; Suhm, M.A.; Eckhoff, M.; Schröder, B.; Mata, R.A. Strained hydrogen bonding in imidazole trimer: A combined infrared, Raman, and theory study. Phys. Chem. Chem. Phys. 2019, 21, 5989–5998. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, L.; Mó, O.; Yáñez, M.; Elguero, J. Very strong hydrogen bonds in neutral molecules: The phosphinic acid dimers. J. Chem. Phys. 1998, 109, 2685–2693. [Google Scholar] [CrossRef]
- DeFord, J.; Chu, F.; Anslyn, E.V. Dimertzation constants for phosphoric acid diesters. Tetrahedron Lett. 1996, 37, 1925–1928. [Google Scholar] [CrossRef]
- Crofts, P.C.; Kosolapoff, G.M. Preparation and determination of apparent dissociation constants of some alkylphosphonic and dialkylphosphinic acids. J. Am. Chem. Soc. 1953, 75, 3379–3383. [Google Scholar] [CrossRef]
- Edmundson, R.S. Dictionary of Organophosphorus Compounds; Chapman and Hall Ltd.: London, UK, 1988. [Google Scholar]
- Evangelisti, L.; Écija, P.; Cocinero, E.J.; Castaño, F.; Lesarri, A.; Caminati, W.; Meyer, R. Proton tunneling in heterodimers of carboxylic acids: A rotational study of the benzoic acid–formic acid bimolecule. J. Phys. Chem. Lett. 2012, 3, 3770–3775. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.A.E.; Suhm, M.A. Vibrational exciton coupling in homo and hetero dimers of carboxylic acids studied by linear infrared and Raman jet spectroscopy. J. Chem. Phys. 2018, 149, 104307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzeli, D.; Petsalakis, I.D.; Theodorakopoulos, G.; Rebek, J., Jr. Encapsulation of monomers, homodimers and heterodimers of amides and carboxylic acids in three non-covalent assemblies. Struct. Chem. 2015, 26, 1585–1601. [Google Scholar] [CrossRef]
- Riccardo Monaco, R.; Poladura, B.; Diaz de Los Bernardos, M.; Leutzsch, M.; Goddard, R.; List, B. Activation of carboxylic acids in asymmetric organocatalysis. Angew. Chem. Int. Ed. 2014, 53, 7063–7067. [Google Scholar] [CrossRef]
- Sharif, S.; Denisov, G.S.; Toney, M.D.; Limbach, H.H. NMR studies of solvent-assisted proton transfer in a biologically relevant Schiff base: Toward a distinction of geometric and equilibrium H-bond isotope effects. J. Am. Chem. Soc. 2006, 128, 3375–3387. [Google Scholar] [CrossRef]
- Kumar, G.A.; McAllister, M.A. Theoretical investigation of the relationship between proton NMR chemical shift and hydrogen bond strength. J. Org. Chem. 1998, 63, 6968–6972. [Google Scholar] [CrossRef]
- Gorobets, N.Y.; Yermolayev, S.A.; Gurley, T.; Gurinov, A.A.; Tolstoy, P.M.; Shenderovich, I.G.; Leadbeater, N.E. Difference between 1H NMR signals of primary amide protons as a simple spectral index of the amide intramolecular hydrogen bond strength. J. Phys. Org. Chem. 2012, 25, 287–295. [Google Scholar] [CrossRef]
- Dohnal, V.; Tkadlecova, M. A simple relation between 1H NMR data and mixing enthalpy for systems with complex formation by hydrogen bonding. J. Phys. Chem. B 2002, 106, 12307–12310. [Google Scholar] [CrossRef]
- Weinhold, F.; Klein, R.A. What is a hydrogen bond? Mutually consistent theoretical and experimental criteria for characterizing H-bonding interactions. Mol. Phys. 2012, 110, 565–579. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Perera, S.A.; Bartlett, R.J. Hydrogen bond types, binding energies, and 1H NMR chemical shifts. J. Phys. Chem. A 1999, 103, 8121–8124. [Google Scholar] [CrossRef]
- Tupikina, E.Y.; Sigalov, M.; Shenderovich, I.G.; Mulloyarova, V.V.; Denisov, G.S.; Tolstoy, P.M. Correlations of NHN hydrogen bond energy with geometry and 1H NMR chemical shift difference of NH protons for aniline complexes. J. Chem. Phys. 2019, 150, 114305. [Google Scholar] [CrossRef] [PubMed]
- Limbach, H.-H.; Tolstoy, P.M.; Perez-Hernandez, N.; Guo, J.; Shenderovich, I.G.; Denisov, G.S. OHO hydrogen bond geometries and NMR chemical shifts: From equilibrium structures to geometric H/D isotope effects with applications for water, protonated water and compressed ice. Israel J. Chem. 2009, 49, 199–216. [Google Scholar] [CrossRef]
- Hansen, P.E.; Spanget-Larsen, J. NMR and IR investigations of strong intramolecular hydrogen bonds. Molecules 2017, 22, 552. [Google Scholar] [CrossRef] [Green Version]
- Ajami, D.; Tolstoy, P.M.; Dube, H.; Odermatt, S.; Koeppe, B.; Guo, J.; Limbach, H.-H.; Rebek Jr., J. Compressed hydrogen bonds isolated in encapsulation complexes. Angew. Chem. Int. Ed. 2011, 50, 528–531. [Google Scholar] [CrossRef]
- Solka, J.L.; Reis, A.H., Jr.; Mason, G.W.; Lewey, S.M.; Peppard, D.F. Di-ϱ-methylphenylphosphoric acid – the solid state structure of a unique dimeric hydrogen bonded phosphoric acid. J. Inorg.Nucl. Chem. 1981, 43, 1451–1464. [Google Scholar]
- Wieczorek, R.; Dannenberg, J.J. H-bonding cooperativity and energetics of α-helix formation of five 17-amino acid peptides. J. Am. Chem. Soc. 2003, 125, 8124–8129. [Google Scholar] [CrossRef]
- Wieczorek, R.; Dannenberg, J.J. Hydrogen-bond cooperativity, vibrational coupling, and dependence of helix stability on changes in amino acid sequence in small 310-helical peptides. A Density Funct. Theory Study. J. Am. Chem. Soc. 2003, 125, 14065–14071. [Google Scholar] [CrossRef]
- Siegel, J.S.; Anet, F.A.I. Dichlorofluoromethane-d: A versatile solvent for VT-NMR experiments. J. Org. Chem. 1988, 53, 2629–2630. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Goodfellow, R.; Granger, P. NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC recommendations 2001). Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
Sample Availability: not available. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | HH | HD |
|---|---|---|
| 1-1a | 13.75 | 13.43 |
| 2-2b | 13.33 | 13.02 |
| 3-3c | n.d. d | n.d. |
| 4-4a | 12.75 | 12.47 |
| 1-2 | 13.80 | 13.69 |
| 1-3 | n.d. | n.d. |
| 1-4 | 14.93 | 15.00 |
| 2-3 | 15.10 | 14.82 |
| 2-4 | 13.74 | n.m. e |
| 3-4 | 15.08 | n.d. |
| Complex | HHH | HHD | HDH | HDD | HHH | HDH | DDH |
|---|---|---|---|---|---|---|---|
| 1-1-1a | - | - | - | - | 14.45 | 14.28 | 14.11 |
| 2-2-2b | - | - | - | - | 13.96 | 13.72 | 13.54 |
| 3-3-3c | - | - | - | - | n.d. d | n.d. | n.d. |
| 4-4-4a | - | - | - | - | 13.76 | 13.54 | 13.38 |
| 1-2-1 | 14.53 | 14.70 | 14.22 | 14.43 | 14.71 | 14.41 | 14.12 |
| 2-1-2 | 14.46 | 14.66 | 14.14 | 14.38 | 14.36 | 14.04 | 13.74 |
| 1-3-1 | 15.74 | n.m. e | n.m. | n.m. | 16.98 | n.m. | n.m. |
| 3-1-3 | 15.94 | n.m. | n.m. | n.m. | 15.98 | n.m. | n.m. |
| 1-4-1 | 14.70 | 15.44 | 14.67 | 15.75 | 16.83 | 16.97 | 16.78 |
| 4-1-4 | 14.43 | 15.07 | 14.46 | 15.48 | 16.76 | 16.94 | 16.74 |
| 2-3-2 | 14.98 | 15.02 | 14.64 | 14.75 | 14.29 | 13.97 | 13.62 |
| 3-2-3 | 14.72 | 14.75 | 14.40 | 14.48 | 15.39 | n.d. | n.d. |
| 2-4-2 | 14.80 | n.m. | n.m. | n.m. | 15.04 | n.m. | n.m. |
| 4-2-4 | 14.81 | n.m. | n.m. | n.m. | 14.88 | n.m. | n.m. |
| 3-4-3 | 14.50 | n.d. | n.d. | n.d. | 15.24 | n.d. | n.d. |
| 4-3-4 | 14.61 | 14.67 | 14.34 | 14.44 | 13.83 | 13.55 | 13.30 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mulloyarova, V.V.; Ustimchuk, D.O.; Filarowski, A.; Tolstoy, P.M. H/D Isotope Effects on 1H-NMR Chemical Shifts in Cyclic Heterodimers and Heterotrimers of Phosphinic and Phosphoric Acids. Molecules 2020, 25, 1907. https://doi.org/10.3390/molecules25081907
Mulloyarova VV, Ustimchuk DO, Filarowski A, Tolstoy PM. H/D Isotope Effects on 1H-NMR Chemical Shifts in Cyclic Heterodimers and Heterotrimers of Phosphinic and Phosphoric Acids. Molecules. 2020; 25(8):1907. https://doi.org/10.3390/molecules25081907
Chicago/Turabian StyleMulloyarova, Valeriia V., Daria O. Ustimchuk, Aleksander Filarowski, and Peter M. Tolstoy. 2020. "H/D Isotope Effects on 1H-NMR Chemical Shifts in Cyclic Heterodimers and Heterotrimers of Phosphinic and Phosphoric Acids" Molecules 25, no. 8: 1907. https://doi.org/10.3390/molecules25081907
APA StyleMulloyarova, V. V., Ustimchuk, D. O., Filarowski, A., & Tolstoy, P. M. (2020). H/D Isotope Effects on 1H-NMR Chemical Shifts in Cyclic Heterodimers and Heterotrimers of Phosphinic and Phosphoric Acids. Molecules, 25(8), 1907. https://doi.org/10.3390/molecules25081907