
Comparison of Substituting Ability of Nitronate versus Enolate for Direct Substitution of a Nitro Group



Abstract
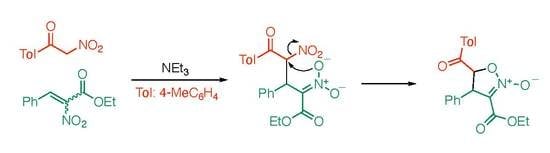
:
1. Introduction
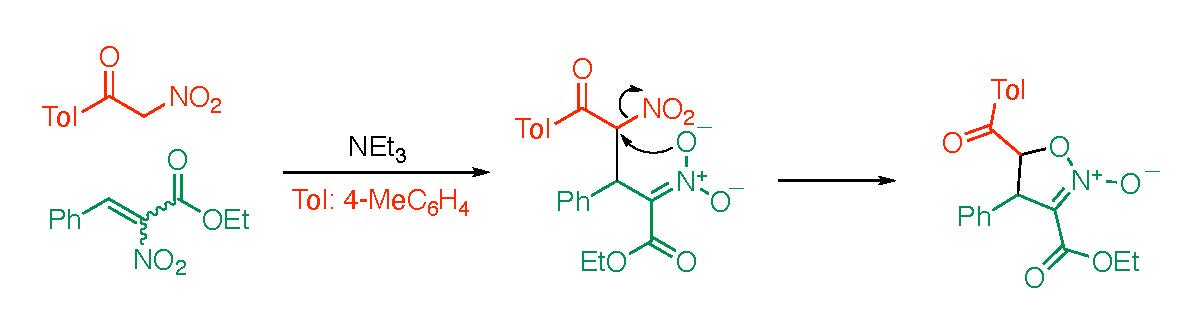
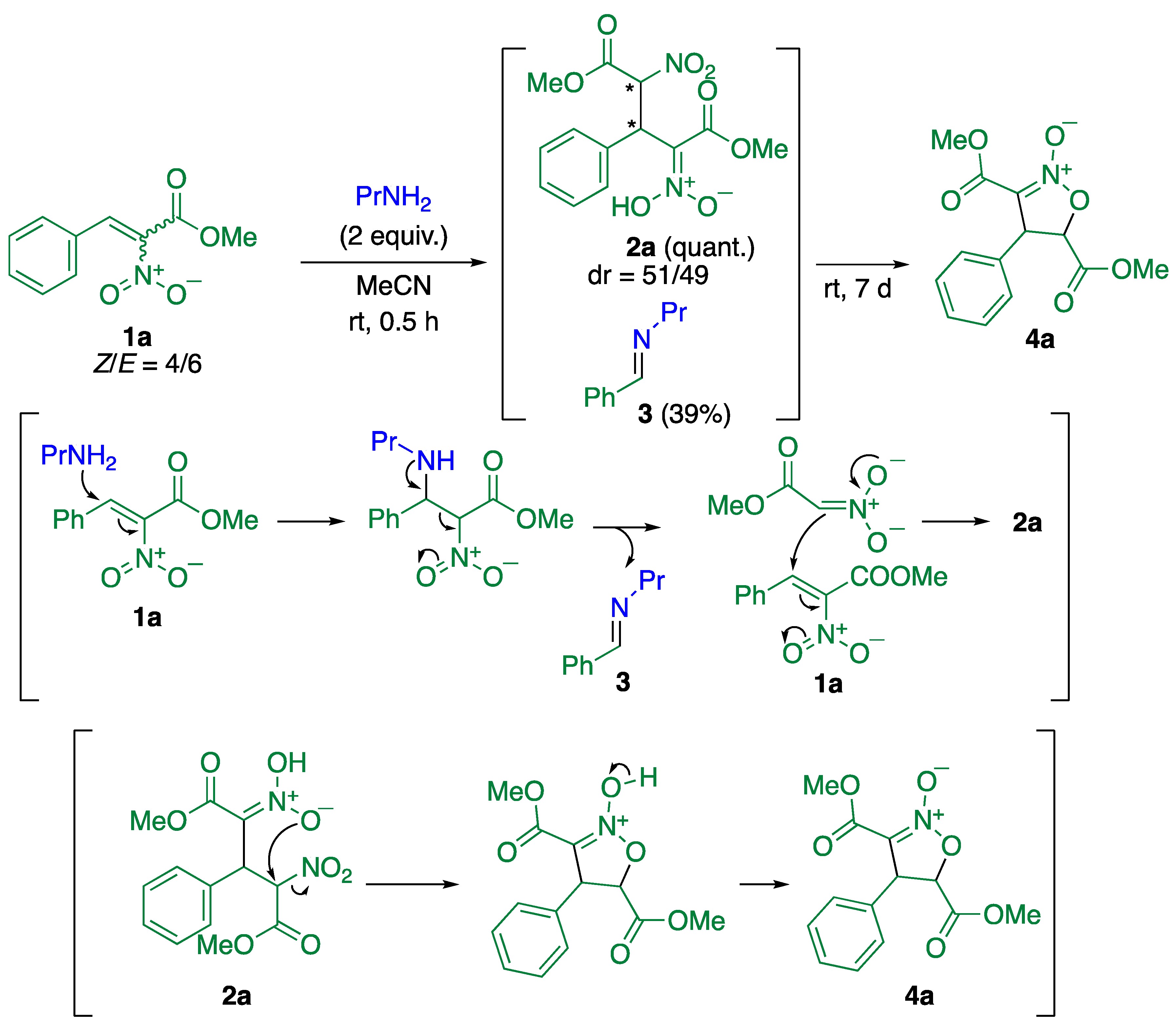
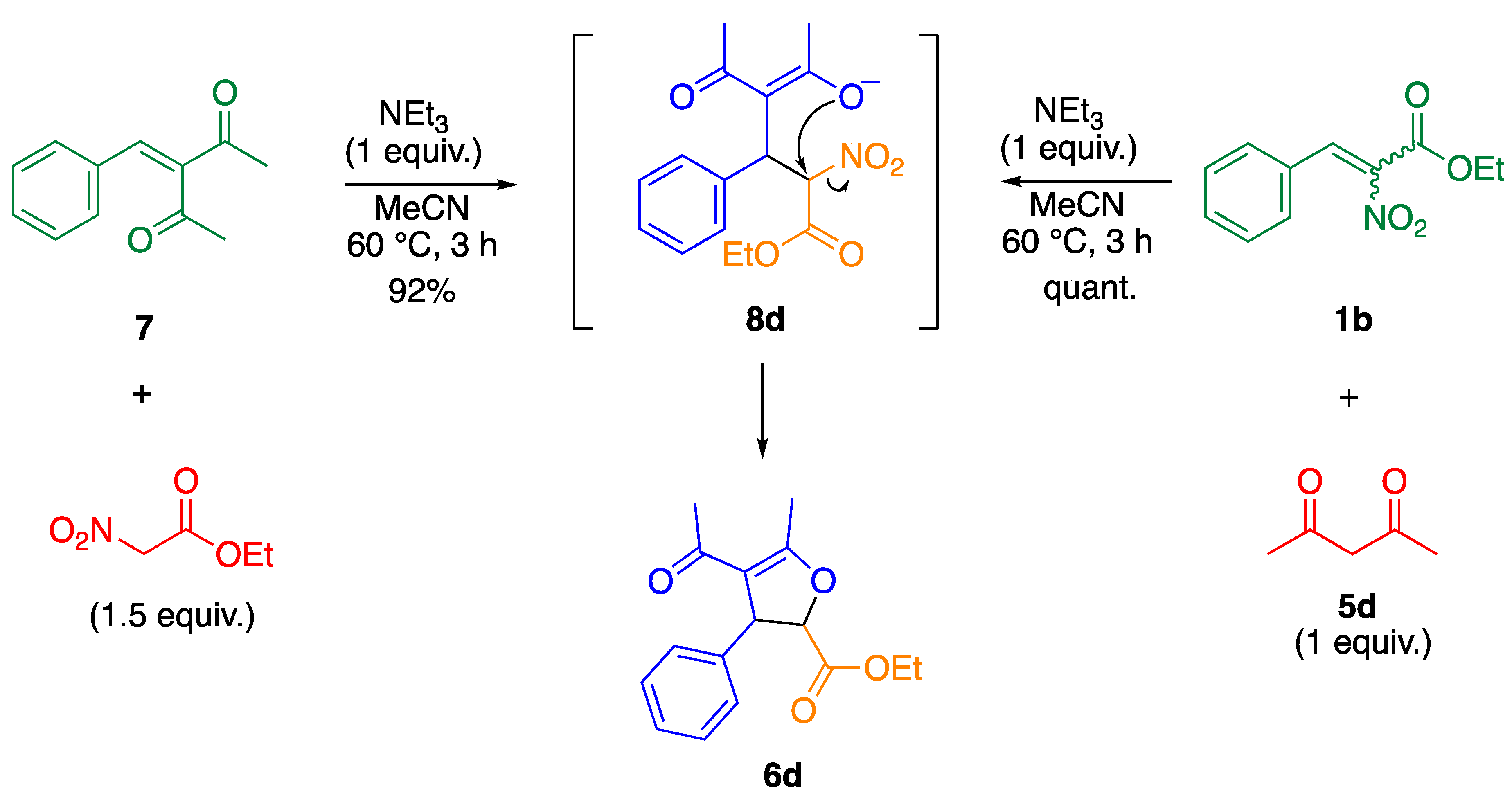
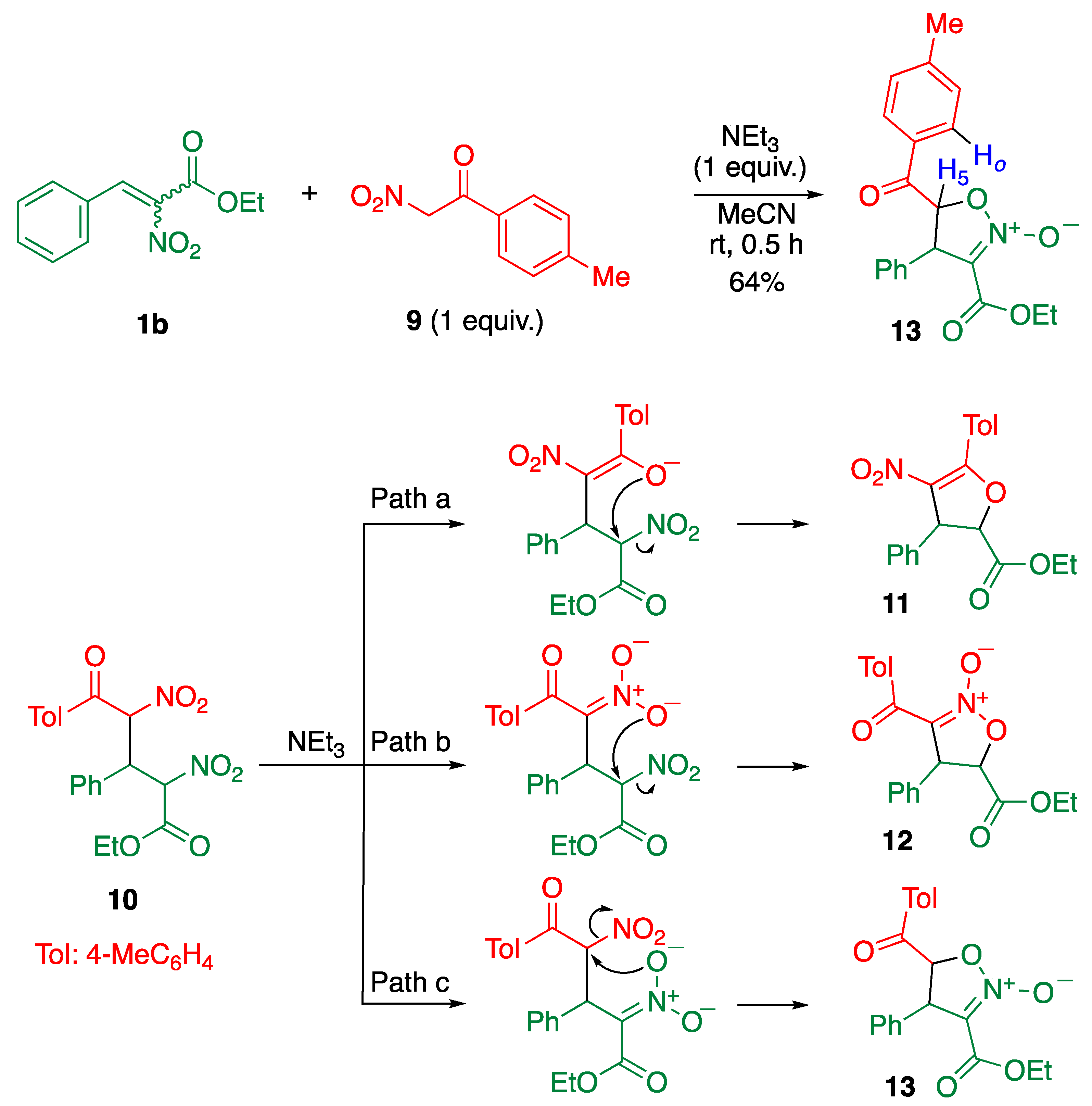
2. Results and Discussion
3. Experimental Section
3.1. General
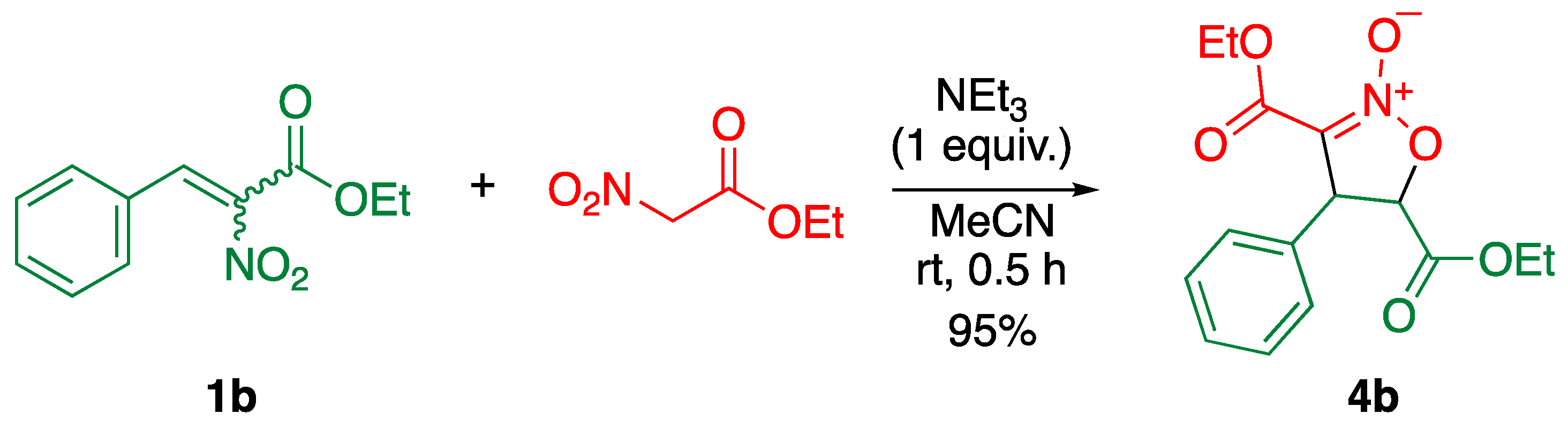
3.2. Synthesis of Isoxazoline N-oxide 4b
3.3. Typical Procedure for Synthesis of 2,3-Dihydrofuran 6
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Asahara, H.; Sofue, A.; Kuroda, Y.; Nishiwaki, N. Alkynylation and cyanation of alkenes using diverse properties of a nitro group. J. Org. Chem. 2018, 83, 13691–13699. [Google Scholar] [CrossRef]
- Nishiwaki, N. Chemistry of nitroquinolones and synthetic application to unnatural 1-methyl-2-quinolone derivatives. Molecules 2010, 15, 5174–5195. [Google Scholar] [CrossRef] [Green Version]
- Hao, F.; Nishiwaki, N. Recent progress in nitro-promoted direct functionalization of pyridones and quinolones. Molecules 2020, 25, 673. [Google Scholar] [CrossRef] [Green Version]
- Chiurchiù, E.; Gabrielli, S.; Ballini, R.; Palmieri, A. A new valuable synthesis of polyfunctionalized furans starting from β-nitroenones and active methylene compounds. Molecules 2019, 24, 4575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, A.; Gabrielli, S.; Ballini, R. Efficient two-step sequence for the synthesis of 2,5-disubstituted furan derivatives from functionalized nitroalkanes: successive amberlyst A21- and amberlyst 15-catalyzed processes. Chem. Commun. 2010, 46, 6165–6167. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, A.; Gabrielli, S.; Parlapiano, M.; Ballini, R. One-pot synthesis of alkyl pyrrole-2-carboxylates starting from β-nitroacrylates and primary amines. RSC Adv. 2015, 5, 4210–4213. [Google Scholar] [CrossRef]
- Mo, Y.; Liu, S.; Liu, Y.; Ye, L.; Shi, Z.; Zhao, Z.; Li, X. Highly stereoselective synthesis of 2,3-dihydrofurans via a cascade michael addition-alkylation process: A nitro group as the leaving group. Chem. Commun. 2019, 55, 6285–6288. [Google Scholar] [CrossRef]
- Noble, A.; Anderson, J.C. Nitro-mannich reaction. Chem. Rev. 2013, 113, 2887–2939. [Google Scholar] [CrossRef]
- Sukhorukov, A.Y. C-H reactivity of the a-position in nitrones and nitronates. Adv. Synth. Catal. 2020, 362, 724–754. [Google Scholar] [CrossRef]
- Baiazitov, R.; Denmark, S.E. Tandem [4 + 2]/[3 + 2] cycloadditions. In Methods and Applications of Cycloaddition Reactions in Organic Syntheses; Nishiwaki, N., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 471–550. [Google Scholar]
- Nishiwaki, N.; Kumegawa, Y.; Iwai, K.; Yokoyama, S. Development of safely handleable synthetic equivalent of cyanonitrile oxide by 1,3-dipolar cycloaddition of nitroacetonitrile. Chem. Commun. 2019, 55, 7903–7905. [Google Scholar] [CrossRef] [Green Version]
- Ballini, R.; Petrini, M. The nitro to carbonyl conversion (nef reaction): New perspectives for a classical transformation. Adv. Synth. Catal. 2015, 357, 2371–2402. [Google Scholar] [CrossRef]
- Iwai, K.; Asahara, H.; Nishiwaki, N. Synthesis of functionalized 3-cyanoisoxazoles using a dianionic reagent. J. Org. Chem. 2017, 82, 5409–5415. [Google Scholar] [CrossRef] [PubMed]
- Kallitsakis, M.G.; Tancini, P.D.; Dixit, M.; Mpourmpakis, G.; Lykakis, I.N. Mechanistic studies on the Michael addition of amines and hydrazines to nitrostyrenes: Nitroalkane elimination via a retro-aza-henry-type process. J. Org. Chem. 2018, 83, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Melot, J.M.; Texier-Boullet, F.; Foucaud, A. Alumina supported potassium fluoride promoted reaction of nitroalkanes with electrophilic alkenes. synthesis of 4,5-dihydrofurans and isoxazoline n-oxides. Tetrahedron 1988, 44, 2215–2224. [Google Scholar] [CrossRef]
- Zhu, C.-Y.; Sun, X.-L.; Deng, X.-M.; Zheng, J.-C.; Tang, Y. Synthesis of isoxazoline N-oxides and its application in the formal synthesis of dehydroclausenamide. Tetrahedron 2008, 64, 5583–5589. [Google Scholar] [CrossRef]
- Zhu, C.-Y.; Deng, X.-M.; Sun, X.-L.; Zheng, J.-C.; Tang, Y. Highly enantioselective synthesis of isoxazoline N-oxides. Chem. Commun. 2008, 738–740. [Google Scholar] [CrossRef]
- Chernysheva, N.B.; Maksimenko, A.S.; Andreyanov, F.A.; Kislyi, V.P.; Strelenko, Y.A.; Khrustalev, V.N.; Victor, V. Synthesis of 3,4-diaryl-5-carboxy-4,5-dihydroisoxazole 2-oxides as valuable synthons for anticancer molecules. Tetrahedron 2017, 73, 6728–6735. [Google Scholar] [CrossRef]
- Le Menn, J.C.; Sarrazin, J.; Tallec, A. Formation of isoxazoline N-oxides and dihydrofurans by cyclocondensation of bromo- or chloromalonate carbanion with michael acceptors. Bull. Soc. Chim. Fr. 1991, 562–565. [Google Scholar]
- Shi, Z.; Tan, B.; Leong, W.; Wen, Y.; Zeng, X.; Lu, M.; Zhong, G. Catalytic asymmetric formal [4 + 1] annulation leading to optically active cis-isoxazoline N-oxides. Org. Lett. 2010, 12, 5402–5405. [Google Scholar] [CrossRef]
- Chen, X.; Peng, Y.; Yu, W.; Zhang, X.; Shao, X.; Xu, X.; Li, Z. Condition-based selective synthesis of 3,4,5-trisubstituted isoxazoline N-oxides, 4,5-dihydroisoxazoles and isoxazoles. ChemistrySelect 2018, 3, 6344–6348. [Google Scholar] [CrossRef]
- Rouf, A.; Sahin, E.; Tanyeli, C. Divergent synthesis of polysubstituted isoxazoles, isoxazoline N-oxides, and dihydroisoxazoles by a one-pot cascade reaction. Tetrahedron 2017, 73, 331–337. [Google Scholar] [CrossRef]
- Zhang, Y.-R.; Luo, F.; Huang, X.-J.; Xie, J.-W. Water-compatible cascade reaction: An efficient route to substituted 2,3-dihydrofurans. Chem. Lett. 2012, 41, 777–779. [Google Scholar] [CrossRef]
- Chuang, C.-P.; Chen, K.-P.; Hsu, Y.-L.; Tsai, A.-I.; Liu, S.-T. α-nitro carbonyl compounds in the synthesis of 2,3-dihydrofurans. Tetrahedron 2008, 64, 7511–7516. [Google Scholar] [CrossRef]
- Berestovitskaya, V.M.; Baichurin, R.I.; Baichuria, L.V.; Fel’gendler, A.V.; Aboskalova, N.I. Geminally activated β-nitrostyrenes in reactions with cyclic β-diketones. Russ. J. Gen. Chem. 2013, 83, 1755–1763. [Google Scholar] [CrossRef]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | R1 | R2 | 5 and 6 | Yield/% |
|---|---|---|---|---|
| 1 | Me | OEt | a | 92 |
| 2 | CF3 | OEt | b | 43 |
| 3 | Ph | OEt | c | 62 |
| 4 | Me | Me | d | quant. |
| 5 | –(CH2)3– | e | quant. | |
| 6 | OEt | OEt | f | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukaijo, Y.; Yokoyama, S.; Nishiwaki, N. Comparison of Substituting Ability of Nitronate versus Enolate for Direct Substitution of a Nitro Group. Molecules 2020, 25, 2048. https://doi.org/10.3390/molecules25092048
Mukaijo Y, Yokoyama S, Nishiwaki N. Comparison of Substituting Ability of Nitronate versus Enolate for Direct Substitution of a Nitro Group. Molecules. 2020; 25(9):2048. https://doi.org/10.3390/molecules25092048
Chicago/Turabian StyleMukaijo, Yusuke, Soichi Yokoyama, and Nagatoshi Nishiwaki. 2020. "Comparison of Substituting Ability of Nitronate versus Enolate for Direct Substitution of a Nitro Group" Molecules 25, no. 9: 2048. https://doi.org/10.3390/molecules25092048
APA StyleMukaijo, Y., Yokoyama, S., & Nishiwaki, N. (2020). Comparison of Substituting Ability of Nitronate versus Enolate for Direct Substitution of a Nitro Group. Molecules, 25(9), 2048. https://doi.org/10.3390/molecules25092048