
Combined Machine Learning and Molecular Modelling Workflow for the Recognition of Potentially Novel Fungicides


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Set
2.2. Data Preprocessing
2.3. Uninformative Variable Elimination Partial-Least Squares Discriminant Analysis (UVE-PLS-DA)
2.4. General Rules for All Regression Procedures in Classification
2.5. Forward Stepwise Multilinear Regression Procedure
2.6. Random Forest Procedures
2.7. UVE-PLS-Random Forest (UVE-PLS-RF)
2.8. Forward Stepwise Limited Correlation Random Forest Procedure
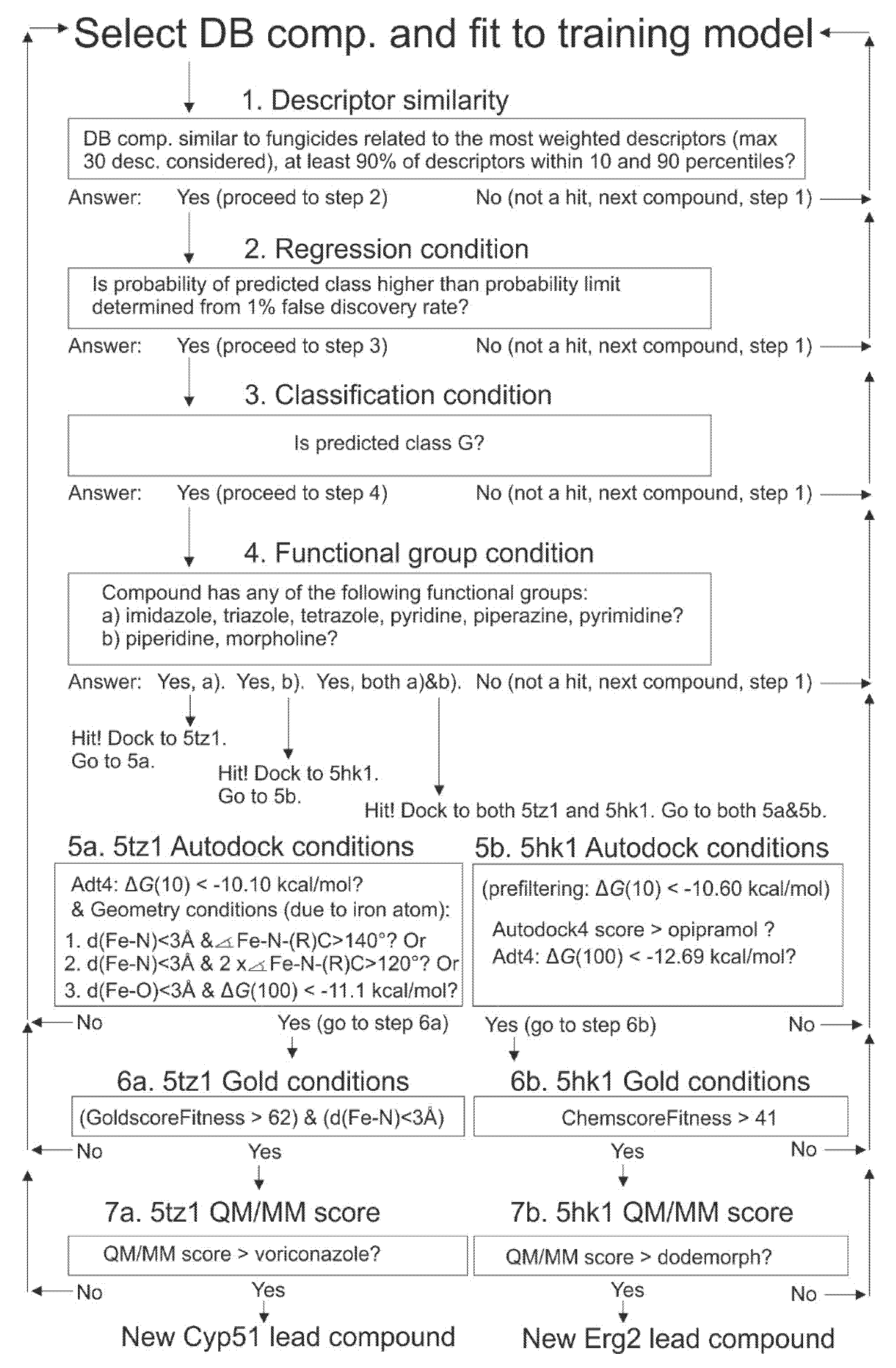
2.9. Selection of Drugbank Hit Compounds, Approach (I)
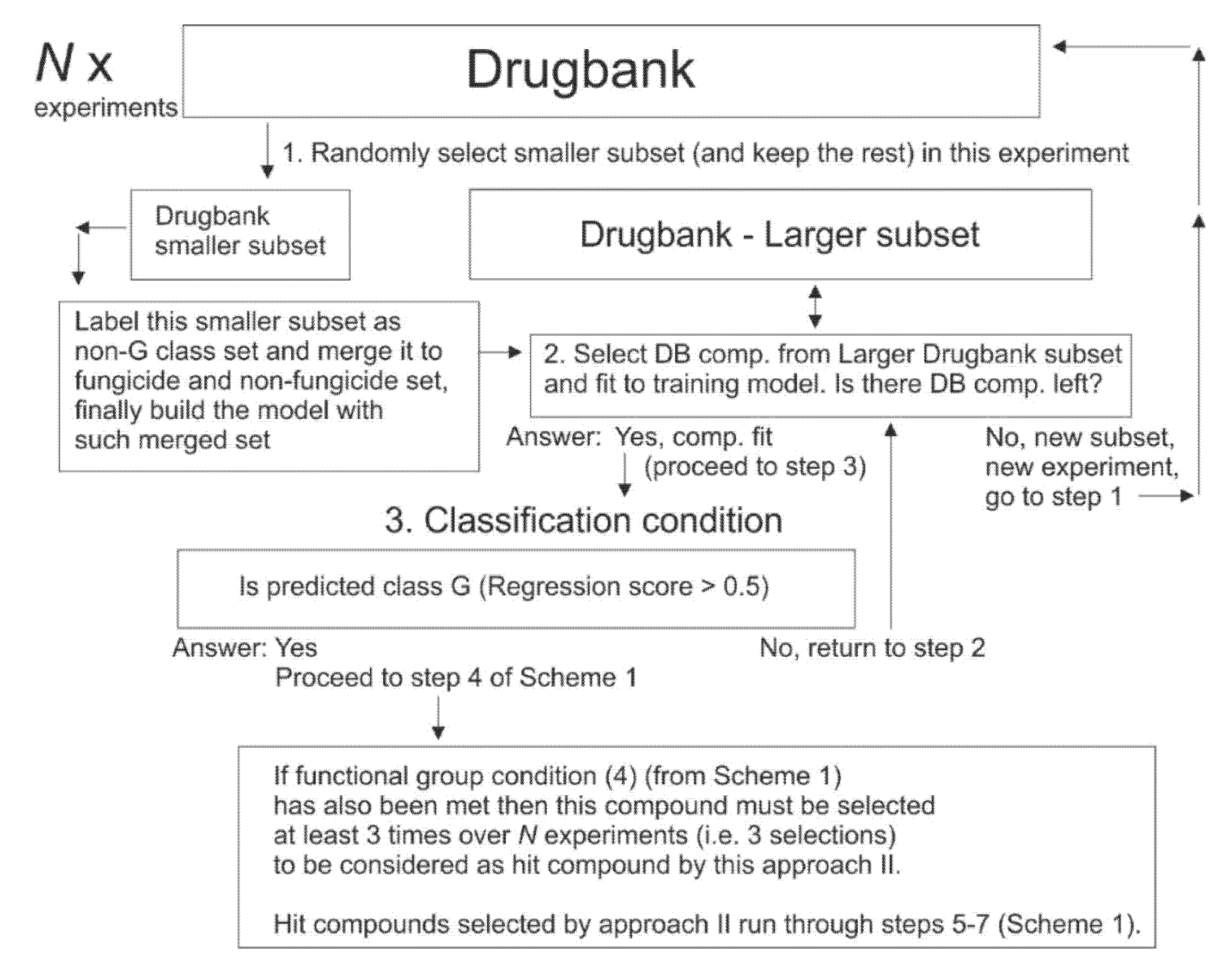
2.10. Selection of Drugbank Hit Compounds, Approach (II)
2.11. Docking Studies
2.11.1. Autodock4 Prefiltering Docking Phase
2.11.2. Docking Phase
2.11.3. Part 3 QM/MM Studies
2.12. Molecular Docking Verifications With Gold Program
3. Results and Discussion
3.1. MOA Classification Approach (I)
3.2. Two-Class Ensemble Regression-Approach (II)
3.3. Docking Study
3.3.1. Autodock Results and Comments
5tz1 Docking
5hk1 Protein
3.3.2. Gold Docking Score Results
3.4. QM/MM Docking and Gold Results
5tz1
3.5. 5hk1 Protein
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fungicide Resistance Action Committee. Available online: http://www.frac.info/ (accessed on 6 June 2019).
- FungiPAD (Fungicide Physicochemical-properties Analysis Database). Available online: http://chemyang.ccnu.edu.cn/ccb/database/FungiPAD/index.php/home (accessed on 6 June 2019).
- Speck-Planche, A.; Kleandrova, V.V.; Rojas-Vargas, J.A. QSAR model toward the rational design of new agrochemical fungicides with a defined resistance risk using substructural descriptors. Mol. Divers. 2011, 15, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Mosbah, A.; Delavenne, E.; Souissi, Y.; Mahjoubi, M.; Jéhan, P.; Le Yondre, N.; Cherif, A.; Bondon, A.; Mounier, J.; Baudy-Floc’h, M.; et al. Novel Antifungal Compounds, Spermine-Like and Short Cyclic Polylactates, Produced by Lactobacillus harbinensis K.V9.3.1Np in Yogurt. Front. Microbiol. 2018, 9, 2252. [Google Scholar] [CrossRef] [Green Version]
- Ogundeji, A.O.; Pohl, C.H.; Sebolai, O.M. The Repurposing of Anti-Psychotic Drugs, Quetiapine and Olanzapine, as Anti-Cryptococcus Drugs. Front. Microbiol. 2017, 8, 815. [Google Scholar] [CrossRef]
- Yu, Q.; Ding, X.; Zhang, B.; Xu, N.; Jia, C.; Mao, J.; Zhang, B.; Xing, L.; Li, M. Inhibitory effect of verapamil on Candida albicans hyphal development, adhesion and gastrointestinal colonization. FEMS Yeast Res. 2014, 14, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Yue, L.; Gu, W.; Li, X.; Zhang, L.; Sun, S. Synergistic Effect of Fluconazole and Calcium Channel Blockers against Resistant Candida albicans. PLoS ONE 2016, 11, e0150859. [Google Scholar] [CrossRef] [PubMed]
- Medoff, G. Reviews of Infectious Diseases. Clin. Infect. Dis. 1983, 5, S614–S619. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Predicting multiple ecotoxicological profiles in agrochemical fungicides: A multi-species chemoinformatic approach. Ecotoxicol. Environ. Saf. 2012, 80, 308–313. [Google Scholar] [CrossRef] [PubMed]
- FRAC Code List 2019: Fungicides Sorted by Mode of Action. Available online: http://www.frac.info/ (accessed on 6 June 2019).
- Hargrove, T.Y.; Friggeri, L.; Wawrzak, Z.; Qi, A.; Hoekstra, W.J.; Schotzinger, R.J.; York, J.D.; Guengerich, F.P.; Lepesheva, G.I. Structural analyses of Candida albicans sterol 14α-demethylase complexed with azole drugs address the molecular basis of azole-mediated inhibition of fungal sterol biosynthesis. J. Biol. Chem. 2017, 292, 6728–6743. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.E.; Warrilow, A.G.; Cools, H.J.; Fraaije, B.A.; Lucas, J.A.; Rigdova, K.; Griffiths, W.J.; Kelly, D.E.; Kelly, S.L. Prothioconazole and Prothioconazole-Desthio Activities against Candida albicans Sterol 14-α-Demethylase. Appl. Environ. Microbiol. 2013, 79, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Brereton, R.G.; Lloyd, G.R. Partial least squares discriminant analysis: Taking the magic away. J. Chemometr. 2014, 28, 213–225. [Google Scholar] [CrossRef]
- Centner, V.; Massart, D.L.; De Noord, O.E.; De Jong, S.; Vandeginste, B.M.; Sterna, C. Elimination of Uninformative Variables for Multivariate Calibration. Anal. Chem. 1996, 68, 3851–3858. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yang, L.; Meng, L.; Wang, J.; Li, S.; Fu, X.; Du, X.; Wu, D. Potential of Visible and Near-Infrared Hyperspectral Imaging for Detection of Diaphania pyloalis Larvae and Damage on Mulberry Leaves. Sensors 2018, 18, 2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Package ‘randomForest’. Available online: https://cran.r-project.org/web/packages/randomForest/randomForest.pdf (accessed on 6 June 2019).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014; Available online: http://www.R-project.org/ (accessed on 3 March 2019).
- Gromek, K.A.; Suchy, F.P.; Meddaugh, H.R.; Wrobel, R.L.; LaPointe, L.; Chu, U.B.; Primm, J.G.; Ruoho, A.E.; Senes, A.; Fox, B.G. The Oligomeric States of the Purified Sigma 1 Receptor are Stabilized by Ligands. J. Biol. Chem. 2014, 289, 20333–20344. [Google Scholar] [CrossRef] [Green Version]
- Laggner, C.; Schieferer, C.; Fiechtner, B.; Poles, G.; Hoffmann, R.D.; Glossmann, H.; Langer, T.; Moebius, F.F. Discovery of High-Affinity Ligands of ó1 Receptor, Erg2, and Emopamil Binding Protein by Pharmacophore Modeling and Virtual Screening. J. Med. Chem. 2005, 48, 4754–4764. [Google Scholar] [CrossRef] [PubMed]
- Chaskar, P.; Zoete, V.; Röhrig, U.F. On-the-Fly QM/MM Docking with Attracting Cavities. J. Chem. Inf. Model. 2017, 57, 73–84. [Google Scholar] [CrossRef]
- ORCA Forum. Available online: https://orcaforum.kofo.mpg.de/app.php/portal (accessed on 11 November 2019).
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456. [Google Scholar] [CrossRef]
- DFT ORCA manual orca manual_4_2_0.pdf. Available online: https://www.afs.enea.it/software/orca/orca_manual_4_2_1.pdf (accessed on 12 December 2019).
- Ferraro, J.R.; Nakamoto, K.; Brown, C.W. Biochemical and Medical Applications. In Introductory Raman Spectroscopy, 2nd ed.; Elsevier Science: San Diego, CA, USA, 2003; pp. 301–302, Chapter 6. [Google Scholar]
- Li, Q.; Gusarov, S.; Evoy, S.; Kovalenko, A. Electronic Structure, Binding Energy, and Solvation Structure of the Streptavidin-Biotin Supramolecular Complex: ONIOM and 3D-RISM Study. J. Phys. Chem. B 2009, 113, 9958–9967. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Warrilow, A.G.; Parker, J.E.; Kelly, D.E.; Kelly, S.L. Azole Affinity of Sterol 14α-Demethylase (CYP51) Enzymes from Candida albicans and Homo sapiens. Antimicrob. Agents Chemother. 2013, 57, 1352–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, A.M.S.; Goldstone, J.V.; Lamb, D.C.; Kubota, A.; Lemaire, B.; Stegeman, J.J. Identification, modeling and ligand affinity of early deuterostome CYP51s, and functional characterization of recombinant zebrafish sterol 14α-demethylase. Biochim. Biophys. Acta 2014, 1840, 1825–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargrove, T.Y.; Wawrzak, Z.; Liu, J.; Waterman, M.R.; Nes, W.D.; Lepesheva, G.I. Structural complex of sterol 14α-demethylase (CYP51) with 14α-methylenecyclopropyl-Δ7-24,25-dihydrolanosterol. J. Lipid Res. 2012, 53, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moebius, F.F.; Reiter, R.J.; Bermoser, K.; Glossmann, H.; Cho, S.Y.; Paik, Y.-K. Pharmacological Analysis of Sterol Δ8-Δ7 Isomerase Proteins with [3H] Ifenprodil. Mol. Pharmacol. 1998, 54, 591–598. [Google Scholar] [CrossRef]
- Metherall, J.E.; Li, H.; Waugh, K. Role of Multidrug Resistance P-glycoproteins in Cholesterol Biosynthesis. J. Biol. Chem. 1996, 271, 2634–2640. [Google Scholar] [CrossRef] [Green Version]
- Moebius, F.F.; Reiter, R.J.; Hanner, M.; Glossmann, H. High affinity of sigma1-binding sites for sterol isomerization inhibitors: Evidence for a pharmacological relationship with the yeast sterol C8–C7 isomerase. Br. J. Pharmacol. 1997, 121, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhao, S.; Kong, X.; Cao, L.; Tian, S.; Ye, Y.; Qiao, C. Design, synthesis and fungicidal evaluation of novel pyraclostrobin analogues. Bioorg. Med. Chem. 2018, 26, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Drugbank. Available online: https://www.drugbank.ca/drugs (accessed on 10 January 2020).
- Velázquez-Libera, J.L.; Rossino, G.; Navarro-Retamal, C.; Collina, S.; Caballero, J. Docking, Interaction Fingerprint, and Three-Dimensional Quantitative Structure—Activity Relationship (3D-QSAR) of Sigma1 Receptor Ligands, Analogs of the Neuroprotective Agent RC-33. Front. Chem. 2019, 7, 496. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No Class. | Method | Nvar | PCC(CV) | PCCte | Passed Step II (III) | No. Hit (Fung) * |
|---|---|---|---|---|---|---|
| 11 | UVE-PLS-DA | 185 | 66.2 | 67.1 | 75 (35) | 34 (13) |
| 11 | UVE-PLS-RF | 185 | 65.0 | 76.8 | 62 (49) | 34 (16) |
| 4 | UVE-PLS-DA | 324 | 77.3 | 77.6 | 636 (2) | 1 (0) |
| 4 | UVE-PLS-RF | 324 | 76.1 | 82.4 | 412 (2) | 2 (2) |
| 3 | UVE-PLS-DA | 115 | 82.8 | 84.1 | 273 (24) | 14 (5) |
| 3 | UVE-PLS-RF | 115 | 81.6 | 86.6 | 334 (20) | 17 (7) |
| 11 | UVE-PLS-FS-MLR | 21 | 68.7 | 63.4 | 64 (34) | 28 (10) |
| 11 | FS-MLR | 30 | 71.8 | 65.9 | 24 (24) | 10 (2) |
| 4 | UVE-PLS-FS-MLR | 6 | 65.7 | 60.8 | 0 (0) | 0 (0) |
| 4 | FS-MLR | 14 | 76.9 | 66.4 | 106 (51) | 24 (5) |
| 3 | UVE-PLS-FS-MLR | 11 | 84.0 | 73.2 | 3 (3) | 0 (0) |
| 3 | FS-MLR | 20 | 91.4 | 79.3 | 100 (18) | 8 (1) |
| No Class. | Corr Gap | Nvar | PCC(CV) | PCCte | Passed Step II (III) | No. Hits (Fung) * |
|---|---|---|---|---|---|---|
| 11 | 0.5 | 16 | 68.1 | 65.9 | 25 (24) | 21 (7) |
| 11 | 0.6 | 14 | 64.4 | 72.0 | 57 (44) | 36 (12) |
| 11 | 0.7 | 22 | 71.8 | 68.3 | 62 (40) | 27 (8) |
| 11 | 0.8 | 17 | 65.6 | 70.7 | 97 (22) | 14 (4) |
| 11 | 0.9 | 14 | 69.9 | 76.8 | 67 (35) | 24 (11) |
| 4 | 0.5 | 17 | 72.9 | 75.2 | 402 (11) | 8 (4) |
| 4 | 0.6 | 13 | 80.1 | 80.0 | 437 (13) | 7 (6) |
| 4 | 0.7 | 18 | 79.7 | 80.0 | 468 (11) | 5 (3) |
| 4 | 0.8 | 17 | 78.5 | 73.6 | 296 (26) | 21 (13) |
| 4 | 0.9 | 18 | 78.5 | 72.8 | 328 (10) | 4 (3) |
| 3 | 0.5 | 15 | 83.4 | 80.5 | 79 (28) | 21 (6) |
| 3 | 0.6 | 19 | 79.8 | 85.4 | 314 (46) | 35 (13) |
| 3 | 0.7 | 12 | 81.6 | 86.6 | 94 (18) | 11 (8) |
| 3 | 0.8 | 18 | 85.3 | 86.6 | 218 (2) | 1 (0) |
| 3 | 0.9 | 16 | 83.4 | 80.5 | 176 (13) | 13 (5) |
| Method | PCC(CV) | PCC(te) | PCCTP(te) | PCC (27 Fung) | Comp. Sel. (Hits) * | Total No. Rep. |
|---|---|---|---|---|---|---|
| UVE-PLS | 98.14 | 98.5 | 71.33 | 77.14 | 42 | 365 |
| UVE-RF | 96.93 | 98.54 | 70.0 | 91.85 | 26 | 148 |
| RF-LM-FS | 98.40 | 98.37 | 66.55 | 90.37 | 78 | 300 |
| All | 97.95 | 98.46 | 69.33 | 85.27 | 100 (37) | 813 |
| ΔG(100) (a) | ΔG(<3Å) (b) | α (c) | ΔG(100) (a) | ΔG(<3Å) (b) | α (c) | ||
|---|---|---|---|---|---|---|---|
| albaconazole | −9.78 | −9.08 | 92.9 | flutriafol | −7.89 | −7.43 | 164.9 |
| bifonazole | −10.02 | −10.02 | 97.0 | fosfluconazole | −10.82 | −7.18 | 157.4 |
| bitertanol | −9.57 | −8.75 | 119.9 | lanoconazole | −8.93 | −8.52 | 106.7 |
| bromuconazole | −8.99 | −8.28 | 168.9 | levoketoconazole | −12.88 | −12.56 | 179.3 |
| butoconazole | −10.14 | −9.51 | 148.10 | luliconazole | −8.97 | −8.88 | 117.1 |
| climbazol | −7.92 | −7.54 | 160.4 | mefentrifluconazole | −9.38 | −8.58 | 115.2 |
| clotrimazole | −9.87 | −9.77 | 157.9 | metconazole | −8.84 | −8.83 | 169.8 |
| croconazole | −9.28 | −8.77 | 170.1 | miconazole | −10.09 | −9.94 | 167.0 |
| eberconazole | −9.98 | −9.98 | 142.6 | myclobutanil | −8.7 | −8.70 | 134.3 (d) |
| econazole | −9.9 | −9.90 | 169.7 | neticonazole | −7.61 | −7.28 | 166.0 |
| efinaconazole | −8.15 | −7.66 | 160.3 | nuarimol | −8.58 | −8.53 | 147.2 |
| epoxiconazole | −9.4 | −8.88 | 164.1 | omoconazole | −10.18 | −10.18 | 169.7 |
| etaconazole | −8.51 | −7.78 | 163.4 | oxiconazole | −10.78 | −10.78 | 168.8 |
| fenarimol | −9.28 | −8.98 | 154.3 | oxpoconazole | −10.17 | −9.99 | 174.4 |
| fenbuconazole | −9.94 | −9.83 | 159.5 | pefurazoate | −7.81 | −7.16 | 110.8 |
| fenticonazole | −11.97 | −11.97 | 164.7 | penconazole | −7.76 | −7.26 | 166.8 |
| fluconazole | −6.76 | −6.76 | 170.4 | posaconazole | −13.81 | −13.04 | 140.9 |
| fluquinconazole | −8.98 | −8.54 | 143.3 | prochloraz | −8.14 | −7.77 | 122.6 |
| furconazole-cis | -8.15 | −8.15 | 117.1 | propiconazole | −8.64 | −8.40 | 148.0 |
| hexaconazole | −7.68 | −6.98 | 174.6 | prothioconazole | −8.12 | −7.98 | 150.2 (e) |
| imazalil | −7.73 | −7.73 | 178.7 | ravuconazole | −10.69 | −9.97 | 171.9 |
| imibenconazole | −9.66 | −9.46 | 154.5 | sertaconazole | −10.6 | −10.6 | 129.0 |
| ipconazole | −9.26 | −8.92 | 176.9 | sulconazole | −9.76 | −9.76 | 162.4 |
| ipfentrifluconazole | −9.53 | −9.53 | 124.0 | tebuconazole | −8.44 | −7.30 | 145.9 |
| isavuconazole | −11.14 | −10.22 | 162.3 | tetraconazole | −7.4 | −6.53 | 170.6 |
| isoconazole | −10.02 | −9.30 | 147.4 | tioconazole | −9.16 | −9.16 | 134.4 |
| itraconazole | −13.8 | −12.52 | 146.1 | triadimenol | −7.98 | −7.36 | 138.7 |
| ketoconazole | −12.59 | −12.59 | 174.7 | triticonazole | −9.64 | −8.37 | 153.1 |
| cyproconazole | −7.72 | −7.72 | 166.6 | voriconazole | −7.75 | −7.75 | 170.0 |
| difenoconazole | −11.09 | −11.09 | 158.8 | oteseconazole | −10.39 | −10.24 | 152.4 (f) |
| diniconazole−M | −8.7 | −7.41 | 109.7 | Average | −9.43 | −9.01 | 150.8 |
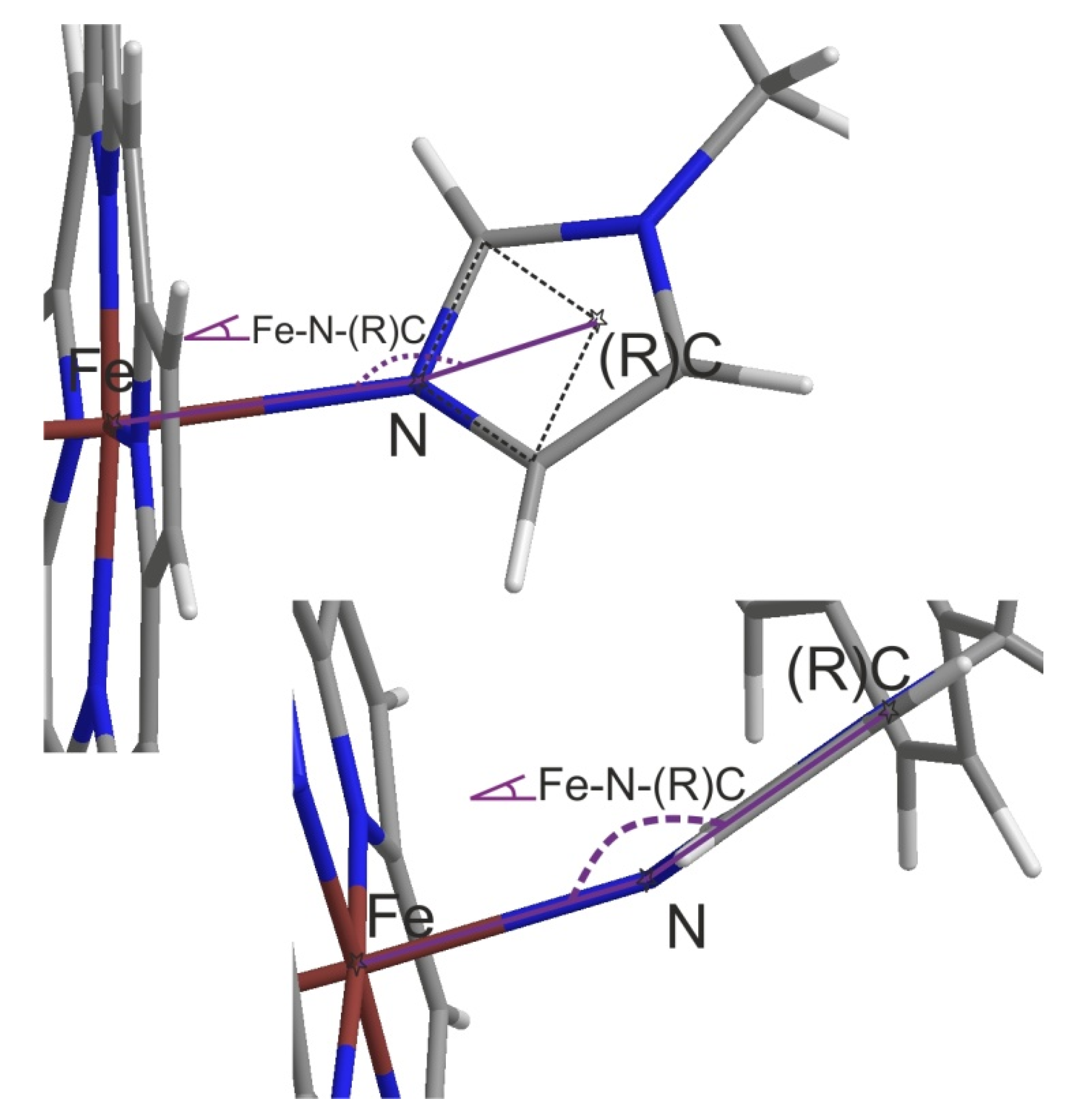
| Hit Comp. | Comment, Default Fe-N<3Å Con-Taining, Otherwise as Commented | For QM/MM | Hit Comp. | Comment, Default Fe-N<3Å Con-Taining, Otherwise as Commented | For QM/MM |
|---|---|---|---|---|---|
| DB12623 | No Fe-N, but with Fe-O, E −16.1 | Yes | DB04107 | No Fe-N, but with Fe-O, E −10.4 | No |
| DB08387 | max Fe-N-®C of 99.5° | No | DB04600 | 2 × max Fe®(R)C: 125°, 123° | Yes |
| DB00354 | No dist(Fe-X) < 3Å (X=N or O) | No | DB07227 | max Fe-N-C of 146.7 + mode ** | Yes × 2 |
| DB08746 | No Fe-N, but with Fe-O, E −10.8 | No | DB06021 | No dist(Fe-X) < 3Å (X=N or O) | No |
| DB12345 | max -N-(R)C of 153.7° | Yes | DB12218 | max Fe-N-(R)C of 147.6° | Yes |
| DB08745 | No Fe-N, but with Fe-O, E −16.1 | Yes | DB02917 | No Fe-N, but with Fe-O, E −10.6 | No |
| DB07578 | max Fe-N-(R)C of 142.3° | Yes | DB12640 | No dist(Fe-X) < 3.5Å (X = any!) | No |
| DB12561 | max Fe-N-(R)C of 119.8° | No | DB00699 | No dist(Fe-X) < 3Å (X=N or O) | No |
| DB12017 | Fe-O, E -11.16, and vina result * | Yes × 2 | DB13113 | max Fe-N-(R)C of 141.4° | Yes |
| DB11679 | No Fe-N, but with Fe-O, E −11.0 | No | DB00737 | No dist(Fe-X) < 3Å (X=N or O) | No |
| DB06834 | No dist(Fe-X) < 3Å (X=N or O) | No | DB07255 | No Fe-N, but with Fe-O, E −9.7 | No |
| DB08922 | No Fe-N, but with Fe-O, E −10.1 | No | DB12364 | No dist(Fe-X) < 3.5Å (X = any!) | No |
| DB04591 | max Fe-N-(R)C of 156.1° | Yes | DB01149 | No dist(Fe-X) < 3Å (X=N or O) | No |
| DB12644 | max Fe-N-(R)C of 114.6° | No | DB07008 | max Fe-N-(R)C of 145.7° | Yes |
| DB04960 | No dist(Fe-X) < 3Å (X=N or O) | No | DB02491 | No Fe-N, but with Fe-O, E −10.2 | No |
| DB13083 | max Fe-N-(R)C of 156.2° | Yes | DB09195 | max Fe-N-(R)C 137.1° (only) | No |
| DB12963 | No dist(Fe-X) < 3.5Å (X = any!) | No | DB07011 | max Fe-N-(R)C of 149.3° | Yes |
| DB04957 | No dist(Fe-X) < 3Å (X=N or O) | No | DB02706 | No Fe-N, but with Fe-O, E −15.0 | Yes |
| DB07878 | No Fe-N, but with Fe-O, E −11.0 | No | DB08560 | No Fe-N, but with Fe-O, E −10.5 | No |
| DB12682 | max Fe-N-(R)C of 153.8° | Yes |
| Compound | ΔG/kcal/mol | Compound | ΔG/kcal/mol |
|---|---|---|---|
| clotrimazole (fungicide) | −37.60 (78) (d) | DB07008 | −124.67 * |
| fluconazole (fungicide) | −46.37 (54) (d) | DB12623 | −71.59 |
| miconazole (fungicide) | −91.74 (79) (d) | DB07578 | −70.49 |
| ketoconazole (fungicide) | −86.58 (85) (d) | DB12017 (b) | −70.17 |
| oteseconazole (fungicide) | −106.36 (98) (d) | DB04591 | −82.30 * |
| voriconazole (fungicide) | −74.92 (84) (d) | DB07011 | −77.96 * |
| water molecule (H2O) | −12.22 | DB08745 | −57.15 |
| DB13083 | −82.73 * | DB13113 | −31.15 |
| DB04600 | −225.38 (a) | DB12345 | −96.20 * |
| DB07227 (b) | −128.79 * | DB12218 | - (c) |
| DB02706 | −59.96 | DB12682 | −115.59 * |
| Docked Comp. | No. Inter. | List of Interacting Amino-Acid Residues |
|---|---|---|
| voriconazole (fung) | 283 | Tyr118, Leu121, Thr122, Phe126, Ile131, Tyr132, Phe228, Gly303, Ile304, Gly307, Gly308, Thr311, Leu376, Ser378, Ile379, Met508 |
| oteseconazole (fung) | 383 | Tyr64, Gly65, Tyr118, Leu121, Thr122, Phe126, Ile131, Tyr132, Phe228, Pro230, Phe233, Gly303, Ile304, Met306, Gly307, Thr311, Leu376, His377, Ser378, Phe380, Tyr505, Ser506, Ser507, Met508 |
| DB13083 (lead) | 406 | Tyr118, Leu121, Thr122, Phe126, Ile131, Tyr132, Phe228, Pro230, Phe233, Gly303, Ile304, Gly307, Gly308, Thr311, Leu376, His377, Ser378, Ile379, Phe380, Ser507, Met508 |
| DB07227 (lead) | 354 | Tyr64, Gly65, Leu87, Leu88, Tyr118, Leu121, Tyr132, Phe228, Pro230, Phe233, Gly307, His310, Thr311, Leu376, His377, Ser378, Ile379, Phe380, Tyr505, Ser506, Ser507, Met508, Val509 |
| DB07008 (lead) | 312 | Tyr118, Leu121, Tyr132, Phe228, Pro230, Phe233, Gly307, Gly308, Thr311, Leu376, His377, Ser378, Ile379, Phe380, Tyr505, Ser506, Ser507, Met508, Val509 |
| DB12345 (lead) | 477 | Tyr64, Gly65, Tyr118, Leu121, Thr122, Tyr132, Phe228, Pro230, Phe233, Gly307, Thr311, Leu376, His377, Ser378, Ile379, Phe380, Tyr505, Ser506, Ser507, Met508 |
| DB04591 (lead) | 442 | Tyr64, Tyr118, Leu121, Thr122, Phe126, Ile131, Tyr132, Phe228, Pro230, Phe233, Gly303, Ile304, Gly307, Gly308, Thr311, Pro375, Leu376, His377, Ser378, Phe380, Tyr505, Ser507, Met508, Val509, Val510 |
| DB07011 (lead) | 401 | Tyr64, Gly65, Pro68, Tyr118, Leu121, Tyr132, Phe228, Pro230, Phe233, Gly307, Gly308, His310, Thr311, Leu376, His377, Ser378, Ile379, Phe380, Tyr505, Ser506, Ser507, Met508, Val509 |
| DB12682(lead) | 377 | Ala61, Tyr64, Gly65, Leu87, Leu88, Tyr118, Leu121, Thr122, Phe126, Ile131, Tyr132, Phe228, Pro230, Phe233, Gly303, Ile304, Gly307, Thr311, Leu376, His377, Ile379, Phe380, Tyr505, Ser506, Met508, Val509 |
| Uncharged Species | ΔG/kcal/mol | Compounds at pH 7,4 (Charged) | ΔG/kcal/mol |
|---|---|---|---|
| Pentazocine (l) [20] | +141.41 | dodemorph (fung) (+1) | −294.39 (r) |
| dodemorph (fung) | −54.39 (r) | tamoxifene (l) (+1) | −285.95 |
| opipramol (l) [20] | −55.16 | Pentazocine (l) (+1) | −102.98 |
| PD144418 (l) [37] | −86.53 | PD144418 (l) (+1) | −367.39 |
| tamoxifene (l) [20] | −45.94 | DB06555 (hit) (+1) | −295.03 |
| DB07075 (hit) | −185.13* | DB08622 (hit) (+1) | −319.22 * |
| DB07075 (hit) (a) | −200.68 | DB08746 (hit) (+1) | −370.02 * |
| DB08622 (hit) | −65.06 * | DB02491 (hit) (+1) | −159.12 |
| DB08746 (hit) | −55.11 * | DB00637 (hit) (+1) | −163.40 |
| DB12345 (hit) | −25.24 | ||
| DB00637 (hit) | +16.96 | ||
| DB06555 (hit) | −50.70 | opipramol (l) (+2) | −498.73 (r) |
| DB02491 (hit) | −8.19 | DB07075 (+2) | −519.81 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jović, O.; Šmuc, T. Combined Machine Learning and Molecular Modelling Workflow for the Recognition of Potentially Novel Fungicides. Molecules 2020, 25, 2198. https://doi.org/10.3390/molecules25092198
Jović O, Šmuc T. Combined Machine Learning and Molecular Modelling Workflow for the Recognition of Potentially Novel Fungicides. Molecules. 2020; 25(9):2198. https://doi.org/10.3390/molecules25092198
Chicago/Turabian StyleJović, Ozren, and Tomislav Šmuc. 2020. "Combined Machine Learning and Molecular Modelling Workflow for the Recognition of Potentially Novel Fungicides" Molecules 25, no. 9: 2198. https://doi.org/10.3390/molecules25092198
APA StyleJović, O., & Šmuc, T. (2020). Combined Machine Learning and Molecular Modelling Workflow for the Recognition of Potentially Novel Fungicides. Molecules, 25(9), 2198. https://doi.org/10.3390/molecules25092198