Reducing Ensembles of Protein Tertiary Structures Generated De Novo via Clustering



Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Structures for a Target Protein
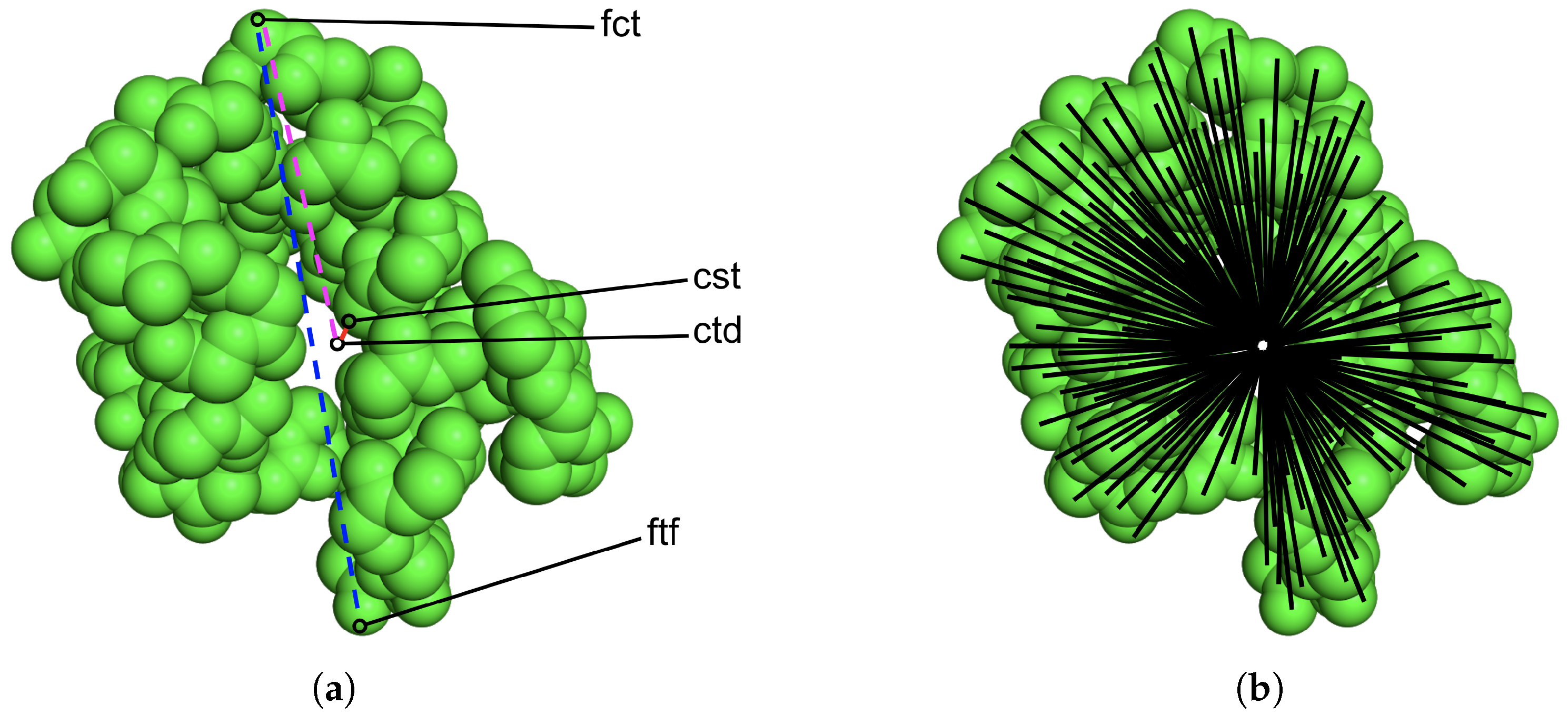
2.2. Stage I: Featurizing Generated Structures
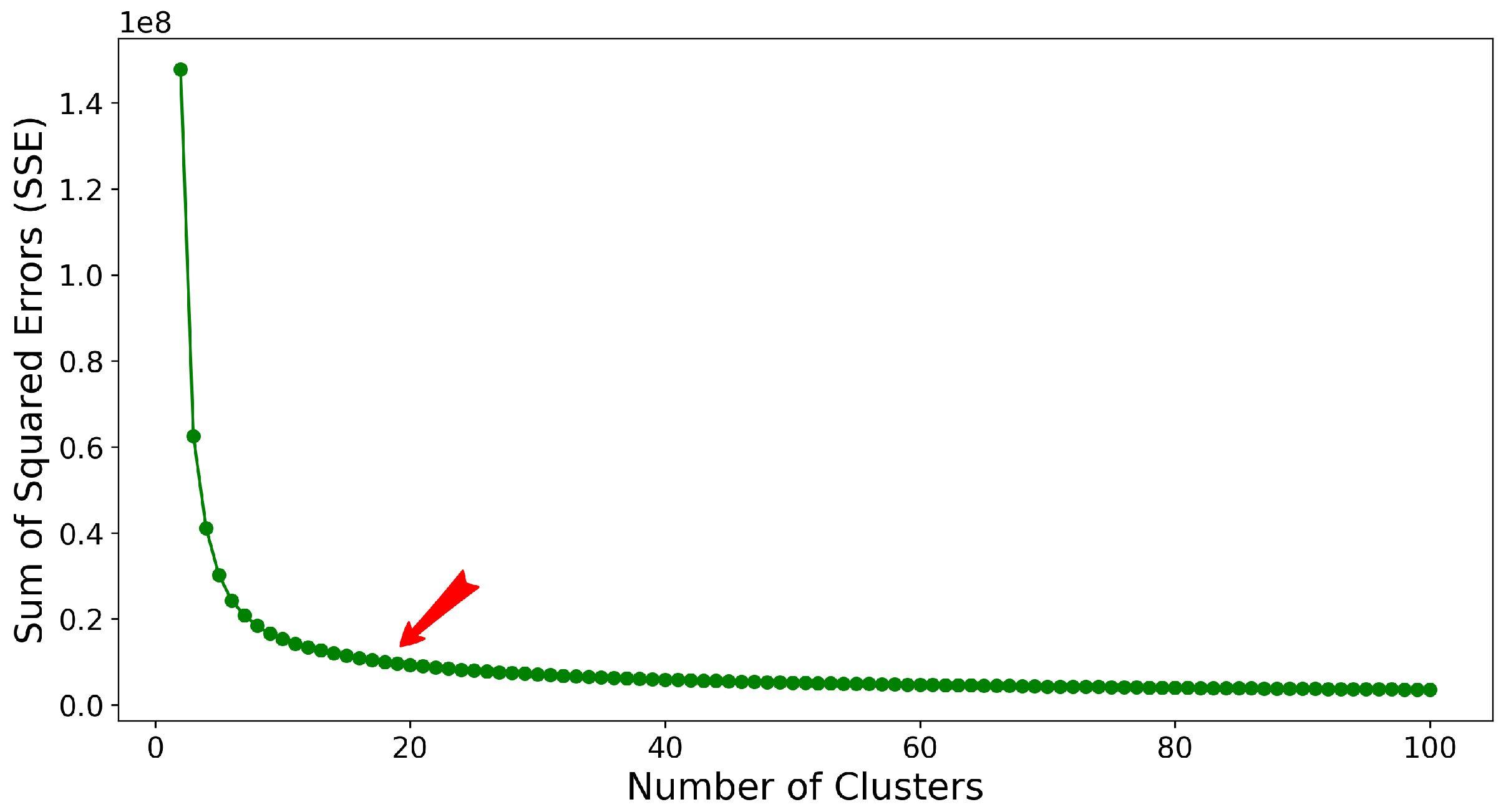
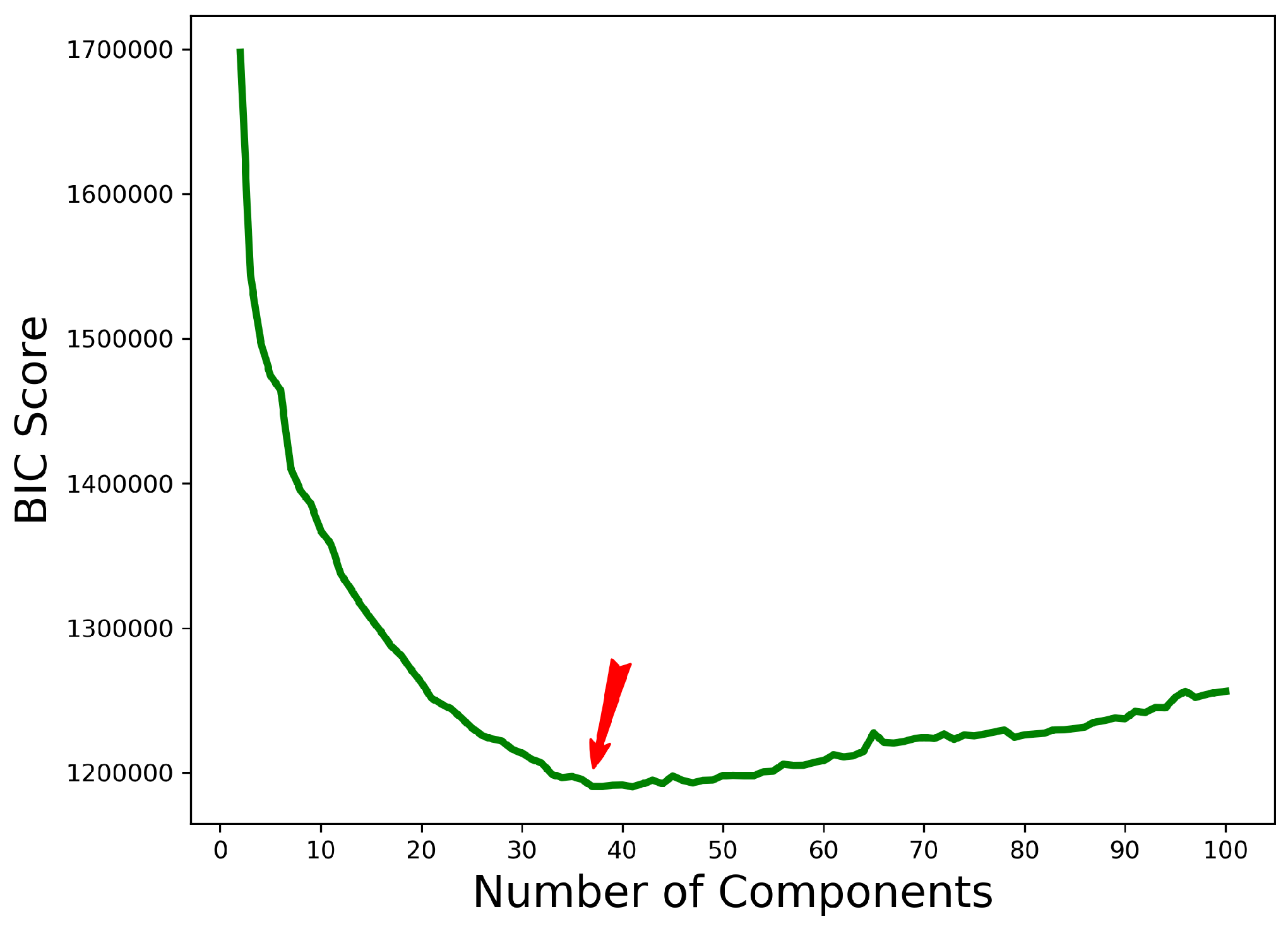
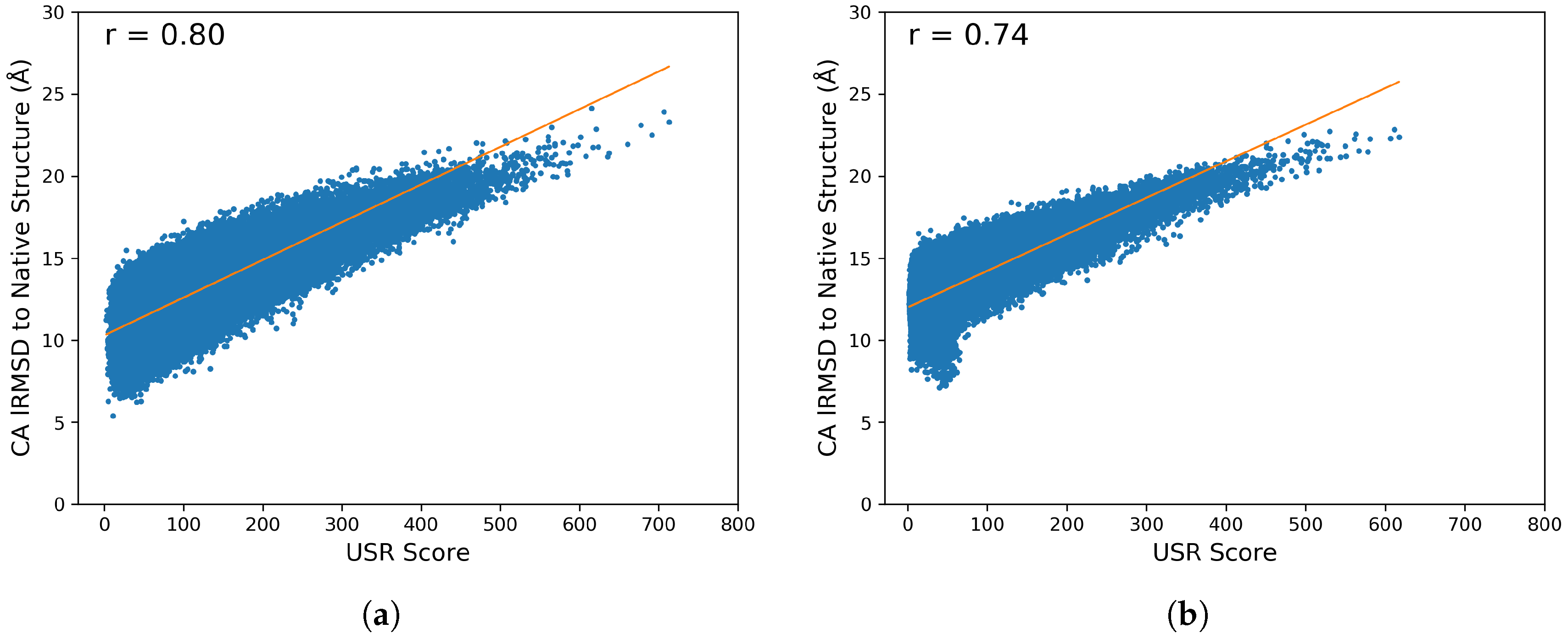
2.3. Stage II: Clustering Featurized Structures
2.4. Stage III: Selecting Structures to Populate the Reduced Ensemble
2.5. Datasets for Evaluation
2.6. Experimental Setup
3. Results
3.1. Comparing Ensemble Sizes Pre- and Post Reduction
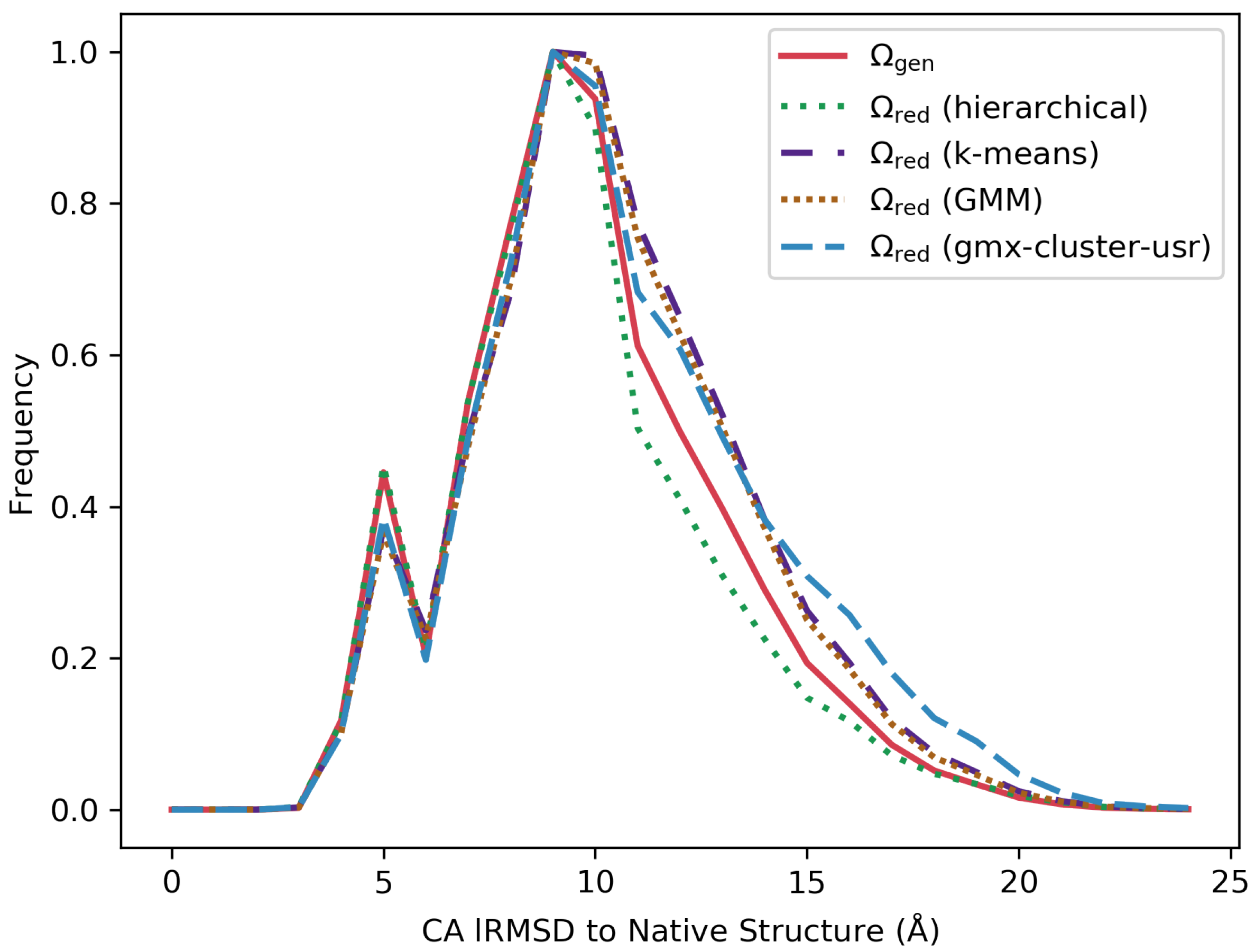
3.2. Comparing Distributions of lRMSDs from the Native Structure Pre- and Post Reduction
3.3. Comparing Distributions of Energies Pre- and Post Reduction
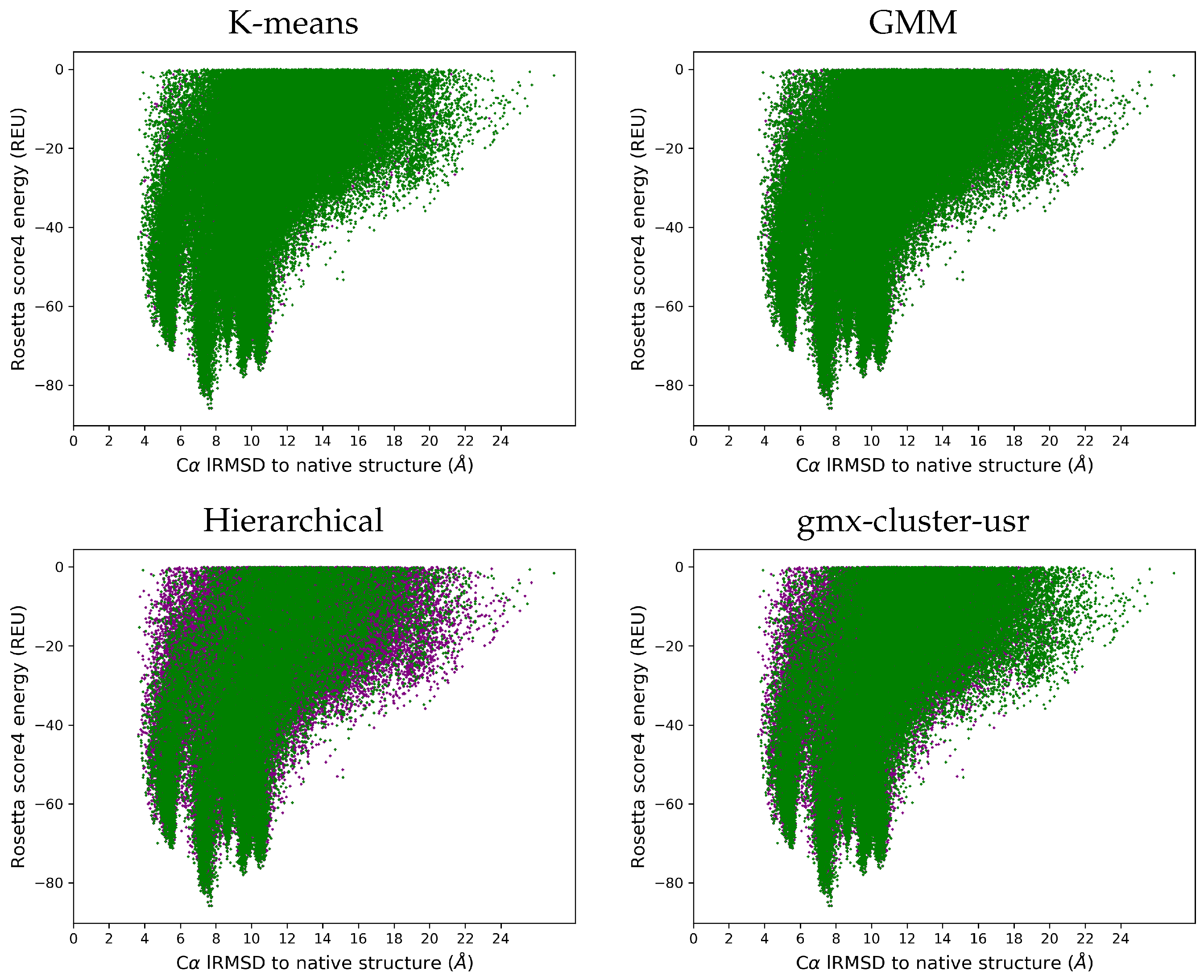
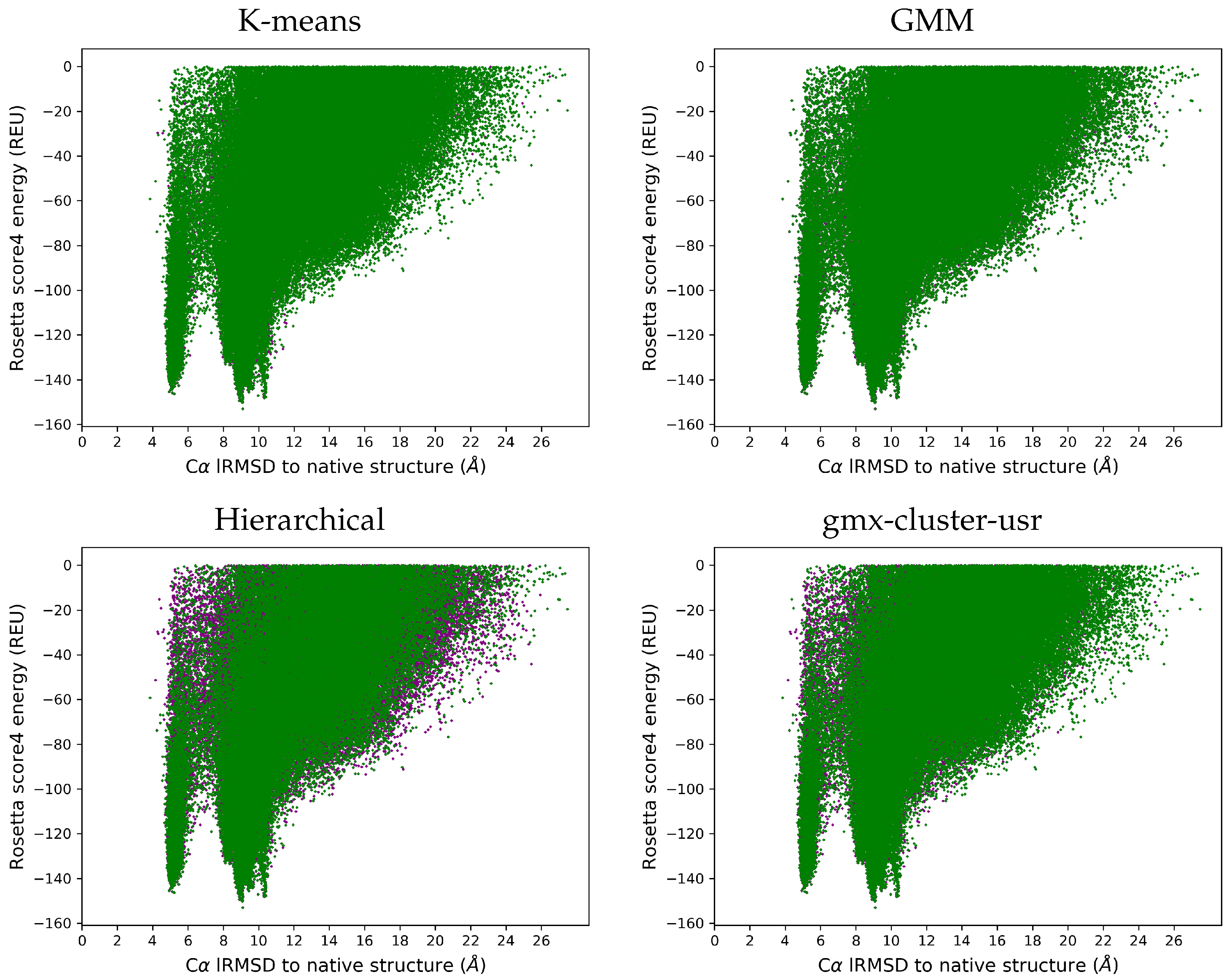
3.4. Visually Comparing Distributions of lRMSDs and Energies Pre- and Post Reduction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BIC | Bayesian Information Criterion |
| CASP | Critical Assessment of protein Structure Prediction |
| DB | Davies-Bouldin |
| GMM | Gaussian Mixture Model |
| HEA | Hybrid Evolutionary Algorithm |
| lRMSD | least Root-Mean-Squared-Deviation |
| PDB | Protein Data Bank |
| PSP | Protein Structure Prediction |
| SSE | Sum of Squared Errors |
| USR | Ultrafast Shape Recognition |
| GROMACS | GROningen MAchine for Chemical Simulations |
References
- Boehr, D.D.; Wright, P.E. How do proteins interact? Science 2008, 320, 1429–1430. [Google Scholar] [CrossRef] [PubMed]
- Maximova, T.; Moffatt, R.; Ma, B.; Nussinov, R.; Shehu, A. Principles and Overview of Sampling Methods for Modeling Macromolecular Structure and Dynamics. PLoS Comput. Biol. 2016, 12, e1004619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaby-Haas, C.E.; de Crécy-Lagard, V. Mining high-throughput experimental data to link gene and function. Trends Biotechnol. 2013, 29, 174–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Lee, J.; Freddolino, P.; Zhang, Y. Ab initio protein structure prediction. In From Protein Structure to Function with Bioinformatics, 2nd ed.; Rigden, D.J., Ed.; Springer: London, UK, 2017; Chapter 1; pp. 3–35. [Google Scholar]
- Levitt, M. Nature of the protein universe. Proc. Natl. Acad. Sci. USA 2009, 106, 11079–11084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R. Four small puzzles that Rosetta doesn’t solve. PLoS ONE 2011, 6, e20044. [Google Scholar] [CrossRef] [Green Version]
- Molloy, K.; Saleh, S.; Shehu, A. Probabilistic Search and Energy Guidance for Biased Decoy Sampling in Ab-initio Protein Structure Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2013, 10, 1162–1175. [Google Scholar] [CrossRef]
- Akhter, N.; Qiao, W.; Shehu, A. An Energy Landscape Treatment of Decoy Selection in Template-free Protein Structure Prediction. Computation 2018, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzym. 2011, 487, 545–574. [Google Scholar]
- Xu, D.; Zhang, Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins Struct. Funct. Bioinform. 2012, 80, 1715–1735. [Google Scholar] [CrossRef] [Green Version]
- Zaman, A.; Shehu, A. Balancing multiple objectives in conformation sampling to control decoy diversity in template-free protein structure prediction. BMC Bioinform. 2019, 20, 211. [Google Scholar] [CrossRef]
- Zaman, A.; De Jong, K.A.; Shehu, A. Using Subpopulation EAs to Map Molecular Structure Landscapes. In Proceedings of the Genetic and Evolutionary Computation (GECCO), Prague, Czech Republic, 13–17 July 2019; ACM: New York, NY, USA, 2019; pp. 1–8. [Google Scholar]
- Olson, B.; Shehu, A. Multi-Objective Optimization Techniques for Conformational Sampling in Template-Free Protein Structure Prediction. In Proceedings of the Bioinform and Comp Biol (BICoB), Las Vegas, NV, USA, 24–26 March 2014; pp. 143–148. [Google Scholar]
- Olson, B.; De Jong, K.A.; Shehu, A. Off-Lattice Protein Structure Prediction with Homologous Crossover. In Proceedings of the Genetic and Evolutionary Computation (GECCO), Amsterdam, The Netherlands, 6–10 July 2013; ACM: New York, NY, USA, 2013; pp. 287–294. [Google Scholar]
- Olson, B.; Shehu, A. Multi-Objective Stochastic Search for Sampling Local Minima in the Protein Energy Surface. In Proceedings of the Bioinf and Comp Biol (BCB), Washington, DC, USA, 22–25 September 2013; pp. 430–439. [Google Scholar]
- Zhang, G.; Ma, L.; Wang, X.; Zhou, X. Secondary Structure and Contact Guided Differential Evolution for Protein Structure Prediction. IEEE Trans. Comput. Biol. Bioinform. 2018, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Mortuza, S.M.; He, B.; Wang, Y.; Zhang, Y. Template-based and free modeling of I-TASSER and QUARK pipelines using predicted contact maps in CASP12. Proteins Struct. Funct. Bioinform. 2018, 86, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Zaman, A.; Parthasarathy, P.V.; Shehu, A. Using Sequence-Predicted Contacts to Guide Template-free Protein Structure Prediction. In Proceedings of the Bioinf and Comp Biol (BCB), Niagara Falls, NY, USA, 7–10 September 2019; ACM: Niagara Falls, NY, USA, 2019; pp. 154–160. [Google Scholar]
- Hou, J.; Wu, T.; Cao, R.; Cheng, J. Protein tertiary structure modeling driven by deep learning and contact distance prediction in CASP13. Proteins 2019, 87, 1165–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaarschmidt, J.; Monastyrskyy, B.; Kryshtafovych, A.; Bonvin, A. Assessment of contact predictions in CASP12: Co-evolution and deep learning coming of age. Proteins 2018, 86, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.J.; Yu, W. De novo protein structure prediction using ultra-fast molecular dynamics simulation. PLoS ONE 2018, 13, e0205819. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Zhou, H.; Skolnick, J. DESTINI: A deep-learning approach to contact-driven protein structure prediction. Sci Rep. 2019, 9, 3514. [Google Scholar] [CrossRef] [Green Version]
- Senior, A.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Monastyrskyy, B.; Fidelis, K.; Schwede, T.; Tramontano, A. Assessment of model accuracy estimations in CASP12. Proteins Struct. Funct. Bioinfom. 2017, 86, 345–360. [Google Scholar] [CrossRef]
- Shehu, A. A Review of Evolutionary Algorithms for Computing Functional Conformations of Protein Molecules. In Computer-Aided Drug Discovery; Zhang, W., Ed.; Methods in Pharmacology and Toxicology; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Zhang, Y.; Skolnick, J. SPICKER: A clustering approach to identify near-native protein folds. J. Comput. Chem. 2004, 25, 865–871. [Google Scholar] [CrossRef]
- Akhter, N.; Chennupati, G.; Djidjev, H.; Shehu, A. Decoy Selection for Protein Structure Prediction Via Extreme Gradient Boosting and Ranking. BMC Bioinform. 2020, in press. [Google Scholar]
- Kabir, L.K.; Hassan, L.; Rajabi, Z.; Shehu, A. Graph-based Community Detection for Decoy Selection in Template-free Protein Structure Prediction. Molecules 2019, 24, 854. [Google Scholar] [CrossRef] [Green Version]
- Akhter, N.; Shehu, A. From Extraction of Local Structures of Protein Energy Landscapes to Improved Decoy Selection in Template-free Protein Structure Prediction. Molecules 2018, 23, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shortle, D.; Simons, K.T.; Baker, D. Clustering of low-energy conformations near the native structures of small proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 11158–11162. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Ng, Y. Calibur: A tool for clustering large numbers of protein decoys. BMC Bioinform. 2010, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Li, S.C.; Bu, D.; Li, M. Clustering 100,000 Protein Structure Decoys in Minutes. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Zaman, A.; Kamranfar, P.; Domeniconi, C.; Shehu, A. Decoy Ensemble Reduction in Template-Free Protein Structure Prediction. In Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Niagara Falls, NY, USA, 7–10 September 2019; ACM: Niagara Falls, NY, USA, 2019; pp. 562–567. [Google Scholar]
- Ballester, P.J.; Richards, W.G. Ultrafast shape recognition to search compound databases for similar molecular shapes. J. Comput. Chem. 2007, 28, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Shehu, A. An Ab-initio tree-based exploration to enhance sampling of low-energy protein conformations. In Robotics: Science and Systems V; Trinkle, J., Matsuoka, Y., Castellanos, J.A., Eds.; The MIT Press: Cambridge, MA, USA, 2009; pp. 241–248. [Google Scholar]
- Olson, B.; Shehu, A. Guiding the Search for Native-like Protein Conformations with an Ab-initio Tree-based Exploration. Int. J. Robot. Res. 2010, 29, 1106–1127. [Google Scholar]
- Molloy, K.; Shehu, A. Interleaving Global and Local Search for Protein Motion Computation. In LNCS: Bioinformatics Research and Applications; Harrison, R., Li, Y., Mandoiu, I., Eds.; Springer International Publishing: Norfolk, VA, USA, 2015; Volume 9096, pp. 175–186. [Google Scholar]
- Molloy, K.; Shehu, A. A General, Adaptive, Roadmap-based Algorithm for Protein Motion Computation. IEEE Trans. NanoBiosci. 2016, 2, 158–165. [Google Scholar] [CrossRef]
- Mani, P.; Vazquez, M.; Metcalf-Burton, J.R.; Domeniconi, C.; Fairbanks, H.; Bal, G.; Beer, E.; Tari, S. The Hubness Phenomenon in High-Dimensional Spaces. In Research in Data Sciences; Gasparovic, E., Domeniconi, C., Eds.; Association for Women in Mathematics; Springer: Cham, Switzerland, 2019; Volume 17, pp. 15–45. [Google Scholar]
- Steinbach, M.; Ertöz, L.; Kumar, V. The Challenges of Clustering High Dimensional Data. In New Directions in Statistics Physics; Wille, L.T., Ed.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 273–309. [Google Scholar]
- Domeniconi, C.; Gunopulos, D.; Ma, S.; Yan, B.; Al-Razgan, M.; Papadopoulos, D. Locally adaptive metrics for clustering high dimensional data. Data Min. Knowl. Discov. 2007, 14, 63–97. [Google Scholar] [CrossRef]
- Gmx Cluster-GROMACS 2018. GROMACS User Guide—Gmx Cluster. Available online: Http://manual.gromacs.org/documentation/2018/onlinehelp/gmx-cluster.html (accessed on 1 May 2020).
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Zhao, Q.; Hautamaki, V.; Fränti, P. Knee point detection in BIC for detecting the number of clusters. In Proceedings of the International Conference on Advanced Concepts for Intelligent Vision Systems, Juan-les-Pins, France, 20–24 October 2008; pp. 664–673. [Google Scholar]
- Likas, A.; Vlassis, N.; Verbeek, J.J. The global k-means clustering algorithm. Pattern Recognit. 2003, 36, 451–461. [Google Scholar] [CrossRef] [Green Version]
- McLachlan, G.J.; Basford, K.E. Mixture Models: Inference and Applications to Clustering; M Dekker: New York, NY, USA, 1988; Volume 84. [Google Scholar]
- Geary, D. Mixture Models: Inference and Applications to Clustering. J. R. Stat. Soc. Ser. A 1989, 152, 126–127. [Google Scholar] [CrossRef]
- Beeferman, D.; Berger, A. Agglomerative clustering of a search engine query log. KDD 2000, 2000, 407–416. [Google Scholar]
- Gowda, K.C.; Krishna, G. Agglomerative clustering using the concept of mutual nearest neighbourhood. Pattern Recognit. 1978, 10, 105–112. [Google Scholar] [CrossRef]
- Davies, D.L.; Bouldin, D.W. A Cluster Separation Measure. IEEE Trans. Pattern Anal. Mach. Intell. 1979, PAMI-1, 224–227. [Google Scholar] [CrossRef]
- Zhang, G.J.; Zhou, X.G.; Yu, X.F.; Hao, H.; Yu, L. Enhancing protein conformational space sampling using distance profile-guided differential evolution. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 1288–1301. [Google Scholar] [CrossRef]
- McLachlan, A.D. A mathematical procedure for superimposing atomic coordinates of proteins. Acta Cryst. A 1972, 26, 656–657. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| K-Means | GMM | Hierarchical | Gmx-Cluster-Usr | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PDB Id | Length | Fold | Red. (%) | Red. (%) | Red. (%) | Red. (%) | |||||
| 1ail | 70 | 250 K | |||||||||
| 1bq9 | 53 | 250 K | |||||||||
| 1c8ca | 64 | 250 K | |||||||||
| 1cc5 | 83 | 250 K | |||||||||
| 1dtja | 76 | 250 K | |||||||||
| 1hhp | 99 | 250 K | |||||||||
| 1tig | 88 | 250 K | |||||||||
| 2ezk | 93 | 250 K | |||||||||
| 2h5nd | 123 | 250 K | |||||||||
| 3gwl | 106 | 250 K | |||||||||
| K-Means | GMM | Hierarchical | Gmx-Cluster-Usr | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CASP Id | Length | Red. (%) | Red. (%) | Red. (%) | Red. (%) | |||||
| T0859-D1 | 129 | 250 K | ||||||||
| T0886-D1 | 69 | 250 K | ||||||||
| T0892-D2 | 110 | 250 K | ||||||||
| T0897-D1 | 138 | 250 K | ||||||||
| T0898-D2 | 55 | 250 K | ||||||||
| T0953s1-D1 | 67 | 250 K | ||||||||
| T0953s2-D3 | 93 | 250 K | ||||||||
| T0957s1-D1 | 108 | 250 K | ||||||||
| T0960-D2 | 84 | 250 K | ||||||||
| T1008-D1 | 77 | 250 K | ||||||||
| Minimum lRMSD (Å) | ||||||||||||
| K-Means | GMM | Hierarchical | Gmx-Cluster-Usr | Truncation | ||||||||
| PDB Id | Diff. | Diff. | Diff. | Diff. | Diff. | |||||||
| 1ail | 0 | 0 | 0 | 0 | ||||||||
| 1bq9 | 0 | 0 | ||||||||||
| 1c8ca | 0 | 0 | 0 | 0 | ||||||||
| 1cc5 | 0 | 0 | 0 | |||||||||
| 1dtja | 0 | 0 | 0 | 0 | ||||||||
| 1hhp | 11 | 11 | 0 | 11 | 0 | |||||||
| 1tig | 0 | 0 | ||||||||||
| 2ezk | 0 | 0 | 0 | 0 | ||||||||
| 2h5nd | 0 | 0 | 0 | 0 | ||||||||
| 3gwl | 0 | 0 | 0 | 0 | ||||||||
| Average lRMSD (Å) | ||||||||||||
| K-Means | GMM | Hierarchical | Gmx-Cluster-Usr | |||||||||
| PDB Id | Diff. | Diff. | Diff. | Diff. | ||||||||
| 1ail | ||||||||||||
| 1bq9 | ||||||||||||
| 1c8ca | ||||||||||||
| 1cc5 | ||||||||||||
| 1dtja | ||||||||||||
| 1hhp | ||||||||||||
| 1tig | ||||||||||||
| 2ezk | ||||||||||||
| 2h5nd | ||||||||||||
| 3gwl | ||||||||||||
| Standard Deviation lRMSD (Å) | ||||||||||||
| K-Means | GMM | Hierarchical | Gmx-Cluster-Usr | |||||||||
| PDB Id | Diff. | Diff. | Diff. | Diff. | ||||||||
| 1ail | 0 | |||||||||||
| 1bq9 | ||||||||||||
| 1c8ca | ||||||||||||
| 1cc5 | ||||||||||||
| 1dtja | ||||||||||||
| 1hhp | ||||||||||||
| 1tig | ||||||||||||
| 2ezk | ||||||||||||
| 2h5nd | ||||||||||||
| 3gwl | ||||||||||||
| Minimum lRMSD (Å) | |||||||||||
| K-Means | GMM | Hierarchical | Gmx-Cluster-Usr | Truncation | |||||||
| CASP Id | Diff. | Diff. | Diff. | Diff. | Diff. | ||||||
| T0859-D1 | 0 | 0 | |||||||||
| T0886-D1 | 0 | 0 | |||||||||
| T0892-D2 | 0 | 0 | 0 | ||||||||
| T0897-D1 | 0 | 0 | |||||||||
| T0898-D2 | 0 | 0 | 0 | 0 | |||||||
| T0953s1-D1 | 0 | 0 | |||||||||
| T0953s2-D3 | 0 | 0 | |||||||||
| T0957s1-D1 | 0 | 0 | |||||||||
| T0960-D2 | 0 | 0 | 0 | 0 | |||||||
| T1008-D1 | 0 | 0 | 0 | 0 | |||||||
| Minimum lRMSD (Å) | |||||||||||
| K-Means | GMM | Hierarchical | Gmx-Cluster-Usr | ||||||||
| CASP Id | Diff. | Diff. | Diff. | Diff. | |||||||
| T0859-D1 | |||||||||||
| T0886-D1 | |||||||||||
| T0892-D2 | |||||||||||
| T0897-D1 | |||||||||||
| T0898-D2 | |||||||||||
| T0953s1-D1 | |||||||||||
| T0953s2-D3 | |||||||||||
| T0957s1-D1 | |||||||||||
| T0960-D2 | |||||||||||
| T1008-D1 | |||||||||||
| Minimum lRMSD (Å) | |||||||||||
| K-Means | GMM | Hierarchical | Gmx-Cluster-Usr | ||||||||
| CASP Id | Diff. | Diff. | Diff. | Diff. | |||||||
| T0859-D1 | |||||||||||
| T0886-D1 | |||||||||||
| T0892-D2 | |||||||||||
| T0897-D1 | |||||||||||
| T0898-D2 | |||||||||||
| T0953s1-D1 | 0 | 0 | |||||||||
| T0953s2-D3 | |||||||||||
| T0957s1-D1 | 0 | 0 | |||||||||
| T0960-D2 | |||||||||||
| T1008-D1 | |||||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaman, A.B.; Kamranfar, P.; Domeniconi, C.; Shehu, A. Reducing Ensembles of Protein Tertiary Structures Generated De Novo via Clustering. Molecules 2020, 25, 2228. https://doi.org/10.3390/molecules25092228
Zaman AB, Kamranfar P, Domeniconi C, Shehu A. Reducing Ensembles of Protein Tertiary Structures Generated De Novo via Clustering. Molecules. 2020; 25(9):2228. https://doi.org/10.3390/molecules25092228
Chicago/Turabian StyleZaman, Ahmed Bin, Parastoo Kamranfar, Carlotta Domeniconi, and Amarda Shehu. 2020. "Reducing Ensembles of Protein Tertiary Structures Generated De Novo via Clustering" Molecules 25, no. 9: 2228. https://doi.org/10.3390/molecules25092228
APA StyleZaman, A. B., Kamranfar, P., Domeniconi, C., & Shehu, A. (2020). Reducing Ensembles of Protein Tertiary Structures Generated De Novo via Clustering. Molecules, 25(9), 2228. https://doi.org/10.3390/molecules25092228