
Benefits and Pitfalls of HPLC Coupled to Diode-Array, Charged Aerosol, and Coulometric Detections: Effect of Detection on Screening of Bioactive Compounds in Apples

 , and
, and
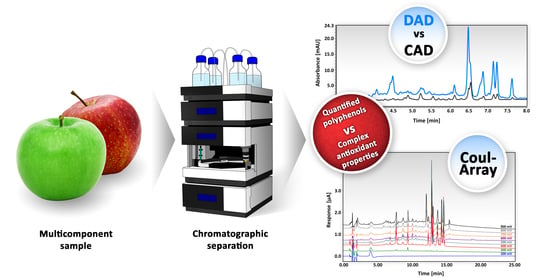
Abstract
:
1. Introduction
2. Results and Discussion
2.1. HPLC Method Development
2.2. HPLC Method Validation
2.2.1. System Suitability
2.2.2. Linearity
2.2.3. Detection and Quantitation Limits
2.2.4. Accuracy, Precision, and Selectivity
2.3. Comparison of Detection Techniques Applied to Apple Extracts Analysis
2.3.1. Sensitivity
2.3.2. HPLC Determination of Selected Bioactive Compounds
2.3.3. Evaluation of Antioxidant Activity
2.3.4. Comparison of Content of Phenolic Compounds and Total Antioxidant Activity of Cultivars
3. Materials and Methods
3.1. Chemicals and Solutions
3.2. Apples and Their Preparation for Analysis
3.3. Chromatography Equipment, Detectors, and Columns
3.4. HPLC Separation Using DAD and CAD
3.5. HPLC Separation with Coulometric Detection
3.6. High-Resolution Mass Spectrometry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cappiello, A.; Famiglini, G.; Palma, P.; Pierini, E.; Termopoli, V.; Trufelli, H. Overcoming matrix effects in liquid chromatography-mass spectrometry. Anal. Chem. 2008, 80, 9343–9348. [Google Scholar] [CrossRef]
- Kang, J.; Hick, L.A.; Price, W.E. Using calibration approaches to compensate for remaining matrix effects in quantitative liquid chromatography/electrospray ionization multistage mass spectrometric analysis of phytoestrogens in aqueous environmental samples. Rapid Commun. Mass Spectrom. 2007, 21, 4065–4072. [Google Scholar] [CrossRef] [Green Version]
- Schuhmacher, J.; Zimmer, D.; Tesche, F.; Pickard, V. Matrix effects during analysis of plasma samples by electrospray and atmospheric pressure chemical ionization mass spectrometry: Practical approaches to their elimination. Rapid Commun. Mass Spectrom. 2003, 17, 1950–1957. [Google Scholar] [CrossRef] [PubMed]
- Avery, M.J. Quantitative characterization of differential ion suppression on liquid chromatography/atmospheric pressure ionization mass spectrometric bioanalytical methods. Rapid Commun. Mass Spectrom. 2003, 17, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.; Wagrowski-Diehl, D.M.; Lu, Z.; Mazzeo, J.R. Systematic and comprehensive strategy for reducing matrix effects in LC/MS/MS analyses. J. Chromatogr. B 2007, 852, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Antignac, J.P.; de Wasch, K.; Monteau, F.; De Brabander, H.; Andre, F.; Le Bizec, B. The ion suppression phenomenon in liquid chromatography–mass spectrometry and its consequences in the field of residue analysis. Anal. Chim. Acta 2005, 529, 129–136. [Google Scholar] [CrossRef]
- Viñas, P.; López-Erroz, C.; Marín-Hernández, J.J.; Hernández-Córdoba, M. Determination of phenols in wines by liquid chromatograpy with photodiode array and fluorescence detection. J. Chromatogr. A 2000, 871, 85–93. [Google Scholar] [CrossRef]
- Hernando, M.D.; Aguera, A.; Fernandez-Alba, A.R. LC-MS analysis and environmental risk of lipid regulators. Anal. Bioanal. Chem. 2007, 387, 1269–1285. [Google Scholar] [CrossRef]
- Ferrer, C.; Lozano, A.; Aguera, A.; Giron, A.J.; Fernandez-Alba, A.R. Overcoming matrix effects using the dilution approach in multiresidue methods for fruits and vegetables. J. Chromatogr. A 2011, 1218, 7634–7639. [Google Scholar] [CrossRef]
- Wu, J.; Qian, X.; Yang, Z.; Zhang, L. Study on the matrix effect in the determination of selected pharmaceutical residues in seawater by solid-phase extraction and ultra-high-performance liquid chromatography-electrospray ionization low-energy collision-induced dissociation tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 1471–1475. [Google Scholar] [PubMed]
- Yarita, T.; Aoyagi, Y.; Otake, T. Evaluation of the impact of matrix effect on quantification of pesticides in foods by gas chromatography-mass spectrometry using isotope-labeled internal standards. J. Chromatogr. A 2015, 1396, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Verdu, C.F.; Gatto, J.; Freuze, I.; Richomme, P.; Laurens, F.; Guilet, D. Comparison of two methods, UHPLC-UV and UHPLC-MS/MS, for the quantification of polyphenols in cider apple juices. Molecules 2013, 18, 10213–10227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walorczyk, S. Validation and use of a QuEChERS-based gas chromatographic-tandem mass spectrometric method for multiresidue pesticide analysis in blackcurrants including studies of matrix effects and estimation of measurement uncertainty. Talanta 2014, 120, 106–113. [Google Scholar] [CrossRef]
- Ciric, A.; Prosen, H.; Jelikic-Stankov, M.; Durdevic, P. Evaluation of matrix effect in determination of some bioflavonoids in food samples by LC-MS/MS method. Talanta 2012, 99, 780–790. [Google Scholar] [CrossRef]
- Buhrman, D.L.; Price, P.I.; Rudewicz, P.J. Quantitation of SR 27417 in human plasma using electrospray liquid chromatography-tandem mass spectrometry: A study of ion suppression. J. Am. Soc. Mass Spectrom. 1996, 7, 1099–1105. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Figares, I.; Rodriguez, L.C.; Gonzalez-Casado, A. Effect of different matrices on physiological amino acids analysis by liquid chromatography: Evaluation and correction of the matrix effect. J. Chromatogr. B 2004, 799, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Caporossi, L.; Tranfo, G.; Paci, E.; Rosa, M.; Capanna, S.; Tidei, F.; Papaleo, B. LC determination of the skin exposure to oxamyl on greenhouse workers and comparison between DAD and MS–MS detection. Chromatographia 2010, 72, 281–287. [Google Scholar] [CrossRef]
- Weingerl, V.; Strlič, M.; Kočar, D. Comparison of methods for determination of polyphenols in wine by HPLC-UV/VIS, LC/MS/MS and spectrophotometry. Acta Chim. Slov. 2009, 56, 698–703. [Google Scholar]
- Lavola, A.; Maukonen, M.; Julkunen-Tiitto, R. Variability in the composition of phenolic compounds in winter-dormant Salix pyrolifolia in relation to plant part and age. Phytochemistry 2018, 153, 102–110. [Google Scholar] [CrossRef]
- Elfalleh, W.; Kirkan, B.; Sarikurkcu, C. Antioxidant potential and phenolic composition of extracts from Stachys tmolea: An endemic plant from Turkey. Ind. Crops Prod. 2019, 127, 212–216. [Google Scholar] [CrossRef]
- Kalili, K.M.; de Villiers, A. Recent developments in the HPLC separation of phenolic compounds. J. Sep. Sci. 2011, 34, 854–876. [Google Scholar] [CrossRef]
- Oyedeji, A.O.; Msagati, T.A.M.; Williams, A.B.; Benson, N.U. Solid-phase extraction and high performance liquid chromatography with diode array detection method for the determination of antibiotic residues in poultry tissues. Chem. Data Collect. 2020, 25, 100312. [Google Scholar] [CrossRef]
- Corell, L.; Armenta, S.; Esteve-Turrillas, F.A.; de la Guardia, M. Flavonoid determination in onion, chili and leek by hard cap espresso extraction and liquid chromatography with diode array detection. Microchem. J. 2018, 140, 74–79. [Google Scholar] [CrossRef]
- Rossle, C.; Wijngaard, H.H.; Gormley, R.T.; Butler, F.; Brunton, N. Effect of storage on the content of polyphenols of minimally processed skin-on apple wedges from ten cultivars and two growing seasons. J. Agric. Food Chem. 2010, 58, 1609–1614. [Google Scholar] [CrossRef]
- Marti, R.; Valcarcel, M.; Herrero-Martinez, J.M.; Cebolla-Cornejo, J.; Rosello, S. Fast simultaneous determination of prominent polyphenols in vegetables and fruits by reversed phase liquid chromatography using a fused-core column. J. Agric. Food Chem. 2015, 169, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Liaudanskas, M.; Viškelis, P.; Kviklys, D.; Raudonis, R.; Janulis, V. A comparative study of phenolic content in apple fruits. Int. J. Food Prop. 2015, 18, 945–953. [Google Scholar] [CrossRef]
- Mihailović, N.R.; Mihailović, V.B.; Kreft, S.; Ćirić, A.R.; Joksović, L.G.; Đurđević, P.T. Analysis of phenolics in the peel and pulp of wild apples (Malus sylvestris (L.) Mill.). J. Food Compos. Anal. 2018, 67, 1–9. [Google Scholar] [CrossRef]
- McGhie, T.K.; Hunt, M.; Barnett, L.E. Cultivar and growing region determine the antioxidant polyphenolic concentration and composition of apples grown in New Zealand. J. Agric. Food Chem. 2005, 53, 3065–3070. [Google Scholar] [CrossRef] [PubMed]
- Plaza, M.; Kariuki, J.; Turner, C. Quantification of individual phenolic compounds’ contribution to antioxidant capacity in apple: A novel analytical tool based on liquid chromatography with diode array, electrochemical, and charged aerosol detection. J. Agric. Food Chem. 2014, 62, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Rodrıguez-Delgado, M.A.; Malovana, S.; Perez, J.P.; Borges, T.; García Montelongo, F.J. Separation of phenolic compounds by high-performance liquid chromatography with absorbance and fluorimetric detection. J. Chromatogr. A 2001, 912, 249–257. [Google Scholar] [CrossRef]
- Granica, S.; Krupa, K.; Klebowska, A.; Kiss, A.K. Development and validation of HPLC-DAD-CAD-MS(3) method for qualitative and quantitative standardization of polyphenols in Agrimoniae eupatoriae herba (Ph. Eur). J. Pharm. Biomed. Anal. 2013, 86, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Tsao, R.; Yang, R.; Young, J.C.; Zhu, H. Polyphenolic profiles in eight apple cultivars using high-performance liquid chromatography (HPLC). J. Agric. Food Chem. 2003, 51, 6347–6353. [Google Scholar] [CrossRef]
- Montero, L.; Herrero, M.; Ibanez, E.; Cifuentes, A. Profiling of phenolic compounds from different apple varieties using comprehensive two-dimensional liquid chromatography. J. Chromatogr. A 2013, 1313, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.; Oliveira, M.B.; Ibanez, E.; Herrero, M. Phenolic profile evolution of different ready-to-eat baby-leaf vegetables during storage. J. Chromatogr. A 2014, 1327, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Bernaldo de Quiros, A.; Lopez-Hernandez, J.; Ferraces-Casais, P.; Lage-Yusty, M.A. Analysis of non-anthocyanin phenolic compounds in wine samples using high performance liquid chromatography with ultraviolet and fluorescence detection. J. Sep. Sci. 2007, 30, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Z.; Xu, X.; Zhou, Q.; Liu, X.; Liao, L.; Zhang, Z.; Wang, Z. Rapid separation and simultaneous quantitative determination of 13 constituents in Psoraleae Fructus by a single marker using high-performance liquid chromatography with diode array detection. J. Sep. Sci. 2017, 40, 4191–4202. [Google Scholar] [CrossRef]
- Haidar Ahmad, I.A.; Blasko, A.; Tam, J.; Variankaval, N.; Halsey, H.M.; Hartman, R.; Regalado, E.L. Revealing the inner workings of the power function algorithm in Charged Aerosol Detection: A simple and effective approach to optimizing power function value for quantitative analysis. J. Chromatogr. A 2019, 1603, 1–7. [Google Scholar] [CrossRef]
- Viinamäki, J.; Ojanperä, I. Photodiode array to charged aerosol detector response ratio enables comprehensive quantitative monitoring of basic drugs in blood by ultra-high performance liquid chromatography. Anal. Chim. Acta 2015, 865, 1–7. [Google Scholar] [CrossRef]
- Dixon, R.W.; Peterson, D.S. Development and testing of a detection method for liquid chromatography based on aerosol charging. Anal. Chem. 2002, 74, 2930–2937. [Google Scholar] [CrossRef]
- Dvořáková, M.; Douanier, M.; Jurková, M.; Kellner, V.; Dostálek, P. Comparison of antioxidant activity of barley (Hordeum vulgare L.) and malt extracts with the content of free phenolic compounds measured by high performance liquid chromatography coupled with CoulArray detector. J. Inst. Brew. 2008, 114, 150–159. [Google Scholar] [CrossRef]
- Kongwong, P.; Morozova, K.; Ferrentino, G.; Poonlarp, P.; Scampicchio, M. Rapid determination of the antioxidant capacity of lettuce by an e-tongue based on flow injection coulometry. Electroanalysis 2018, 30, 230–237. [Google Scholar] [CrossRef]
- Ullucci, P.A.; Acworth, I.N.; Crafts, C.; Bailey, B.A.; Plante, M. Targeted Analyses of Secondary Metabolites in Herbs, Spices, and Beverages Using a Novel Spectro-Electro Array Platform (Application Note No. AN70713_E 07/16S); Thermo Fisher Scientific: Waltham, MA, USA, 2016. [Google Scholar]
- Lima, L.G.B.; Montenegro, J.; Abreu, J.P.; Santos, M.C.B.; Nascimento, T.P.D.; Santos, M.D.S.; Ferreira, A.G.; Cameron, L.C.; Ferreira, M.S.L.; Teodoro, A.J. Metabolite Profiling by UPLC-MS(E), NMR, and Antioxidant Properties of Amazonian Fruits: Mamey Apple (Mammea Americana), Camapu (Physalis Angulata), and Uxi (Endopleura Uchi). Molecules 2020, 25, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Chen, L.; Yu, J.; Cui, L.; Ali, I.; Song, X.; Park, J.H.; Wang, D.; Wang, X. Flavonoid epimers from custard apple leaves, a rapid screening and separation by HSCCC and their antioxidant and hypoglycaemic activities evaluation. Sci. Rep. 2020, 10, 8819. [Google Scholar] [CrossRef] [PubMed]
- Percival, B.C.; Wann, A.; Zbasnik, R.; Schlegel, V.; Edgar, M.; Zhang, J.; Ampem, G.; Wilson, P.; Gresley, A.L.; Naughton, D.; et al. Evaluations of the Peroxidative Susceptibilities of Cod Liver Oils by a 1H NMR Analysis Strategy: Peroxidative Resistivity of a Natural Collagenous and Biogenic Amine-Rich Fermented Product. Nutrients 2020, 12, 753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grootveld, M.; Percival, B.C.; Leenders, J.; Wilson, P.B. Potential Adverse Public Health Effects Afforded by the Ingestion of Dietary Lipid Oxidation Product Toxins: Significance of Fried Food Sources. Nutrients 2020, 12, 974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, F.T.; Remane, D. Aspects of matrix effects in applications of liquid chromatography-mass spectrometry to forensic and clinical toxicology—A review. Anal. Bioanal. Chem. 2012, 403, 2155–2172. [Google Scholar] [CrossRef]
- Sklenářová, H.; Bílková, A.; Pechová, M.; Chocholouš, P. Determination of major phenolic compounds in apples: Part I-optimization of high-performance liquid chromatography separation with diode array detection. J. Sep. Sci. 2018, 41, 3042–3050. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA). Guideline on Bioanalytical Method Validation; European Medicines Agency: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Baker, T.R.; Regg, B.T. A multi-detector chromatographic approach for characterization and quantitation of botanical constituents to enable in silico safety assessments. Anal. Bioanal. Chem. 2018, 410, 5143–5154. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.E.; Liang, L.J.; Yu, X.H.; Wu, H.; Tu, P.F.; Ma, Z.J.; Zhao, K.J. Quality assessment of Astragali Radix from different production areas by simultaneous determination of thirteen major compounds using tandem UV/charged aerosol detector. J. Pharm. Biomed. Anal. 2019, 165, 233–241. [Google Scholar] [CrossRef]
- Dai, J.; Mumper, R.J. Plant phenolics: Extraction, analysis and their antioxidant and anticancer properties. Molecules 2010, 15, 7313–7352. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DAD Detector | tR 1 (min) | Repeatability, RSD (%) | S 3 | RS 4 | PC 5 | Rf 6 | |
|---|---|---|---|---|---|---|---|
| tR | A 2 | ||||||
| Gallic acid 280 nm | 2.81 | 0.15 | 0.19 | 0.91 | - | 49.77 | 0.63 |
| Chlorogenic acid 320 nm | 4.47 | 0.14 | 0.36 | 0.95 | 17.55 | 58.60 | 1.6 |
| Epicatechin 280 nm | 5.19 | 0.17 | 0.50 | 0.82 | 7.12 | 54.61 | 2.02 |
| Rutin 254 nm | 6.09 | 0.15 | 0.16 | 0.97 | 9.36 | 67.78 | 2.54 |
| Phloridzin 280 nm | 7.60 | 0.11 | 0.16 | 0.87 | 15.50 | 45.72 | 3.42 |
| Quercetin 365 nm | 9.57 | 0.11 | 0.51 | 1.20 | 17.93 | 27.61 | 4.56 |
| Phloretin 280 nm | 10.48 | 0.10 | 0.52 | 0.91 | 8.23 | 43.67 | 5.09 |
| CAD Detector | |||||||
| Gallic acid | 2.83 | 0.65 | 0.95 | 0.96 | - | 79.41 | 0.65 |
| Chlorogenic acid | 4.50 | 0.42 | 0.40 | 0.93 | 17.53 | 79.41 | 1.62 |
| Epicatechin | 5.22 | 0.33 | 0.36 | 0.86 | 7.16 | 64.29 | 2.04 |
| Rutin | 6.12 | 0.25 | 0.42 | 0.95 | 9.38 | 67.50 | 2.56 |
| Phloridzin | 7.64 | 0.20 | 0.67 | 0.87 | 15.45 | 61.36 | 3.44 |
| Quercetin | 9.60 | 0.20 | 0.39 | 1.16 | 17.91 | 64.29 | 4.58 |
| Phloretin | 10.51 | 0.16 | 0.09 | 0.91 | 8.27 | 61.36 | 5.11 |
| DAD Detector | LOD (µg/mL) | LOQ (µg/mL) | Calibration Range (µg/mL) | Regression Equation | R2 |
|---|---|---|---|---|---|
| Gallic acid 280 nm | 0.03 | 0.10 | 0.10–20 | 0.2393x − 0.0132 | 0.9989 |
| Chlorogenic acid 320 nm | 0.03 | 0.10 | 0.10–20 | 0.2547x − 0.0298 | 0.9983 |
| Epicatechin 280 nm | 0.07 | 0.25 | 0.25–20 | 0.0876x − 0.0083 | 0.9985 |
| Rutin 365 nm | 0.07 | 0.25 | 0.25–20 | 0.1584x − 0.0092 | 0.9990 |
| Phloridzin 280 nm | 0.03 | 0.10 | 0.10–20 | 0.2174x − 0.0100 | 0.9992 |
| Quercetin 365 nm | 0.03 | 0.10 | 0.10–20 | 0.3971x − 0.0820 | 0.9977 |
| Phloretin 280 nm | 0.03 | 0.10 | 0.10–20 | 0.3791x − 0.0162 | 0.9992 |
| CAD Detector | |||||
| Gallic acid | 0.30 | 1.00 | 1.00–20 | 0.0058x − 0.0024 | 0.9949 |
| Chlorogenic acid | 0.30 | 1.00 | 1.00–20 | 0.0096x − 0.0031 | 0.9943 |
| Epicatechin | 0.30 | 1.00 | 1.00–20 | 0.0163x − 0.0047 | 0.9973 |
| Rutin | 0.30 | 1.00 | 1.00–20 | 0.0145x − 0.0017 | 0.9985 |
| Phloridzin | 0.30 | 1.00 | 1.00–20 | 0.0196x − 0.0019 | 0.9995 |
| Quercetin | 0.30 | 1.00 | 1.00–20 | 0.0224x − 0.0116 | 0.9956 |
| Phloretin | 0.15 | 0.50 | 0.50–20 | 0.0309x − 0.0420 | 0.9996 |
| Analyte/Spiked Level (µg/mL) | Recovery (%) | Intra-Day Precision (%) | |||||
|---|---|---|---|---|---|---|---|
| DAD | |||||||
| 0.2 | 1 | 5 | 10 | 15 | 20 | Cultivar HL 1343 | |
| Gallic acid | 81.7 | 98.5 | 94.3 | 93.7 | 93.6 | 89.3 | |
| Chlorogenic acid | 93.2 | 101.6 | 96.3 | 97.4 | 89.6 | 86.8 | 3.4 |
| Epicatechin | 86.0 | 95.4 | 97.6 | 100.2 | 97.5 | 90.9 | 7.1 |
| Rutin | 94.6 | 104.8 | 100.1 | 102.7 | 97.0 | 92.8 | 3.2 |
| Phloridzin | 100.3 | 97.4 | 92.8 | 94.3 | 93.1 | 88.0 | 1.9 |
| Quercetin | 85.2 | 82.9 | 86.2 | 81.1 | 82.2 | 81.0 | |
| Phloretin | 85.1 | 89.4 | 86.8 | 89.2 | 90.0 | 86.6 | |
| CAD | |||||||
| Gallic acid | 100.9 | 99.7 | 102.0 | 105.8 | 102.3 | 101.4 | |
| Chlorogenic acid | 108.2 | 109.7 | 109.4 | 106.0 | 97.0 | 104.1 | 5.1 |
| Epicatechin | 114.3 | 116.2 | 117.5 | 108.7 | 98.6 | 103.2 | 7.3 |
| Rutin | 93.5 | 97.8 | 98.7 | 90.6 | 92.1 | 92.4 | 7.8 |
| Phloridzin | 88.6 | 97.0 | 89.1 | 92.8 | 88.2 | 88.8 | 6.0 |
| Quercetin | 80.8 | 87.7 | 88.3 | 89.7 | 83.6 | 86.6 | |
| Phloretin | 87.2 | 90.2 | 88.5 | 87.0 | 88.1 | 86.9 | |
| Compound | p-Value |
|---|---|
| Gallic acid | 7.24 × 10−6 |
| Chlorogenic acid | 2.67 × 10−6 |
| Epicatechin | 2.58 × 10−5 |
| Rutin | 6.12 × 10−6 |
| Phloridzin | 1.45 × 10−7 |
| Quercetin | 1.81 × 10−7 |
| Phloretin | 3.05 × 10−7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hollá, M.; Bílková, A.; Jakubec, P.; Košková, S.; Kočová Vlčková, H.; Šatínský, D.; Švec, F.; Sklenářová, H. Benefits and Pitfalls of HPLC Coupled to Diode-Array, Charged Aerosol, and Coulometric Detections: Effect of Detection on Screening of Bioactive Compounds in Apples. Molecules 2021, 26, 3246. https://doi.org/10.3390/molecules26113246
Hollá M, Bílková A, Jakubec P, Košková S, Kočová Vlčková H, Šatínský D, Švec F, Sklenářová H. Benefits and Pitfalls of HPLC Coupled to Diode-Array, Charged Aerosol, and Coulometric Detections: Effect of Detection on Screening of Bioactive Compounds in Apples. Molecules. 2021; 26(11):3246. https://doi.org/10.3390/molecules26113246
Chicago/Turabian StyleHollá, Marcela, Aneta Bílková, Pavel Jakubec, Stanislava Košková, Hana Kočová Vlčková, Dalibor Šatínský, František Švec, and Hana Sklenářová. 2021. "Benefits and Pitfalls of HPLC Coupled to Diode-Array, Charged Aerosol, and Coulometric Detections: Effect of Detection on Screening of Bioactive Compounds in Apples" Molecules 26, no. 11: 3246. https://doi.org/10.3390/molecules26113246
APA StyleHollá, M., Bílková, A., Jakubec, P., Košková, S., Kočová Vlčková, H., Šatínský, D., Švec, F., & Sklenářová, H. (2021). Benefits and Pitfalls of HPLC Coupled to Diode-Array, Charged Aerosol, and Coulometric Detections: Effect of Detection on Screening of Bioactive Compounds in Apples. Molecules, 26(11), 3246. https://doi.org/10.3390/molecules26113246