
CuI-Catalyzed Coupling Reactions of 4-Iodopyrazoles and Alcohols: Application toward Withasomnine and Homologs

 ,
,
Abstract
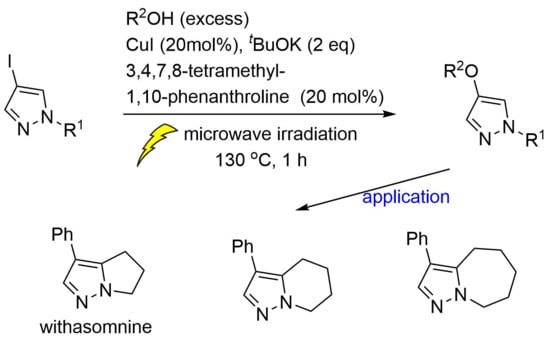
:
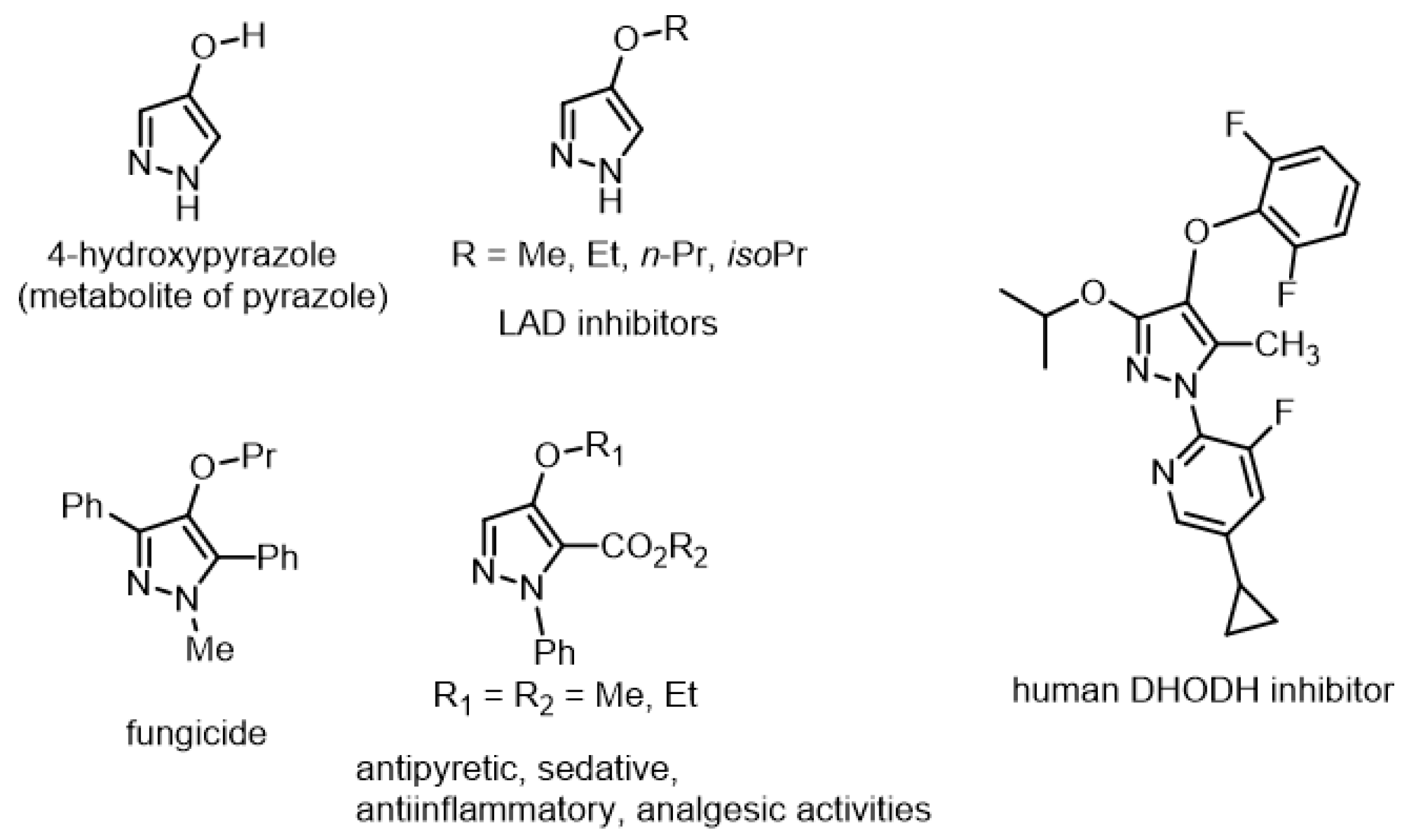
1. Introduction
2. Results and Discussions
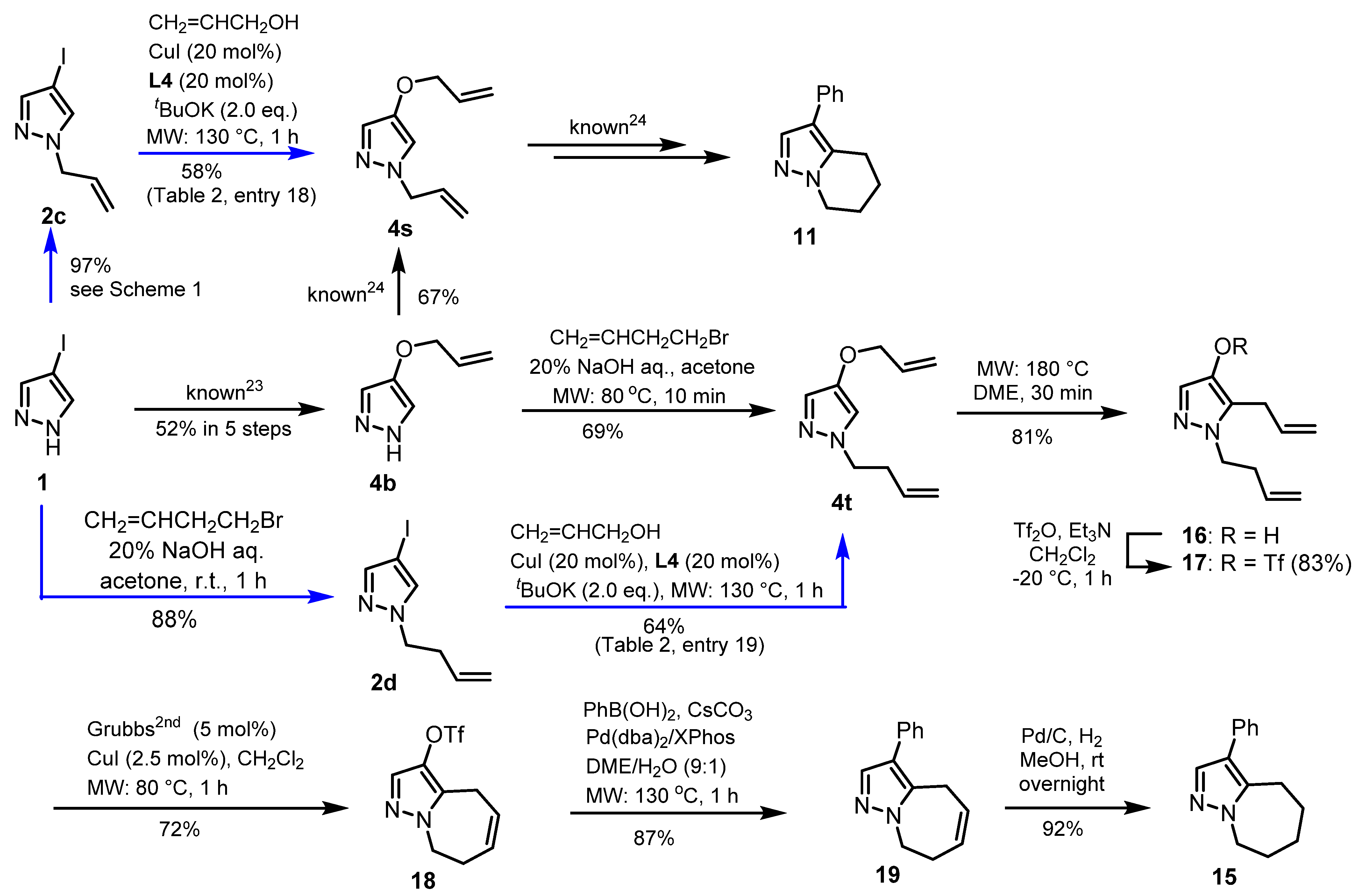
2.1. Investigation of 4-O-Allylation of 4-Iodopyrazole
2.2. C4-Alkoxylation of 4-iodopyrazole with Alcohols Using CuI-Catalyzed Coupling
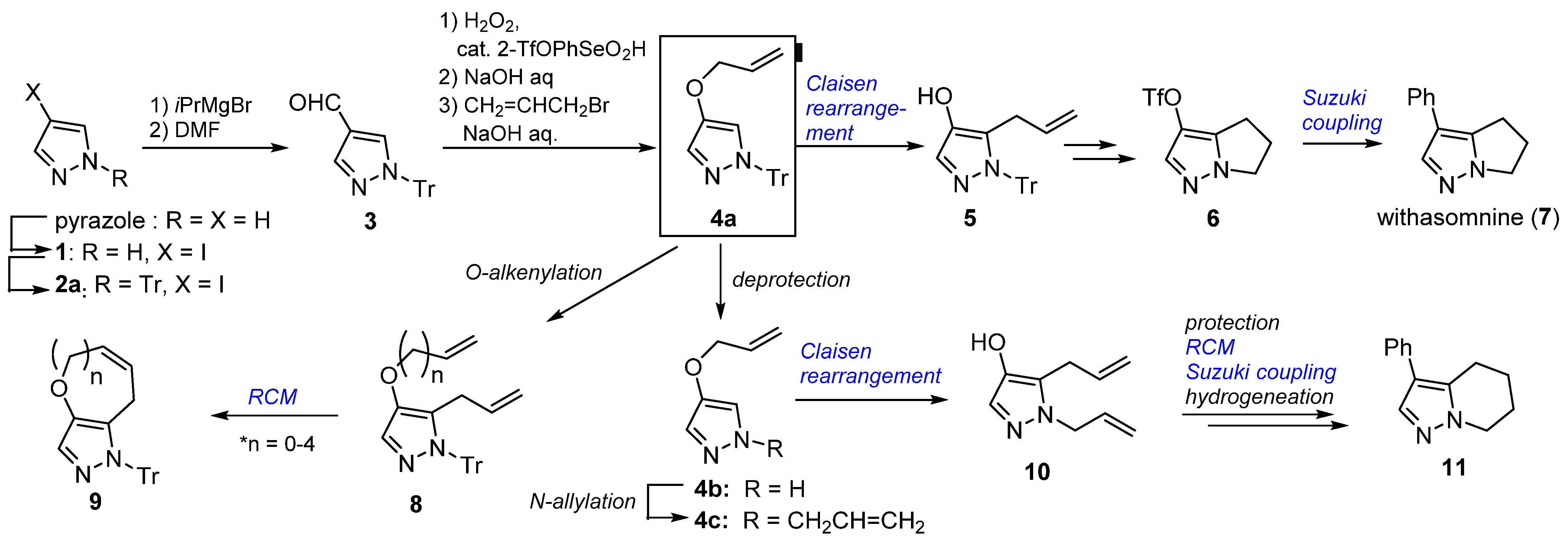
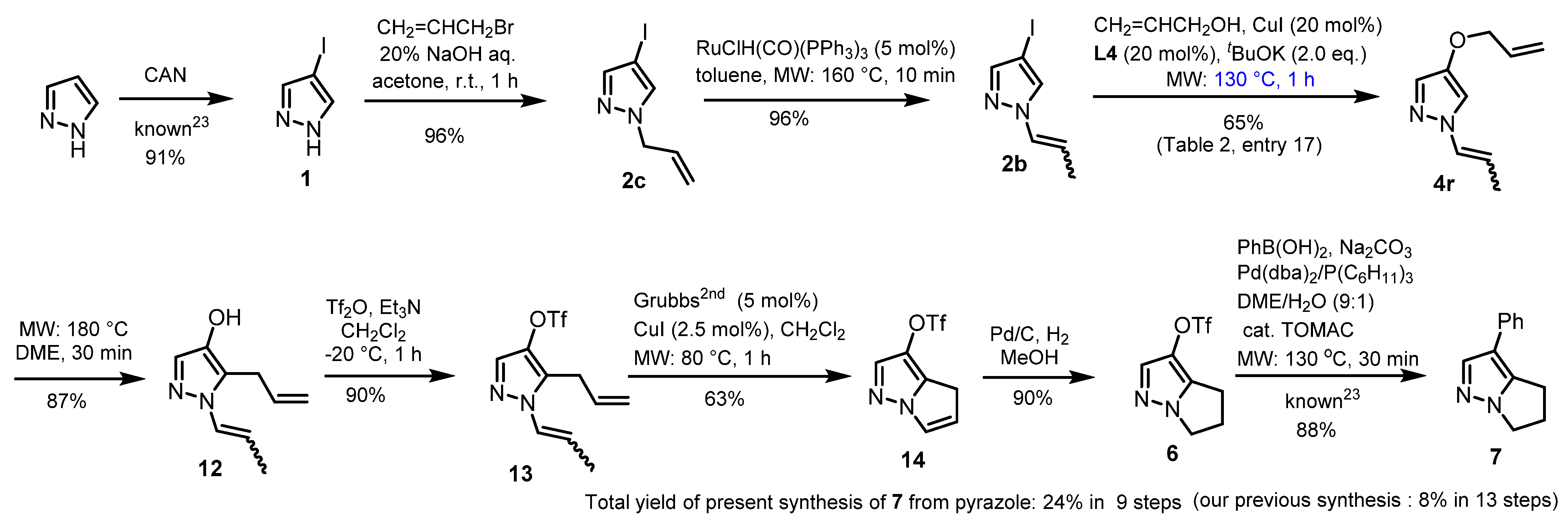
2.3. Application to Improved Synthesis of Withasomnine and Six- and Seven-Membered Cyclic Homologs
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. CuI-Catalyzed Coupling Reactions of 4-iodo-1H-1-tritylpyrazole with Alcohols (Table 1 and Table 2)
- 4s: known [24]
4.3. Modified Synthesis of Withasomnine, (Scheme 2)
4.3.1. Synthesis of 1-allyl-4-iodo-1H-pyrazole (2c)
4.3.2. Synthesis of (E/Z)-4-iodo-1-(prop-1-en-1-yl)-1H-pyrazole (2b)
4.3.3. Synthesis of (E/Z)-5-allyl-1-(prop-1-en-1-yl)-1H-pyrazol-4-yl trifluoromethane-sulfonate (12)
4.3.4. Synthesis of (E/Z)-5-allyl-1-(prop-1-en-1-yl)-1H-pyrazol-4-yl trifluoromethanesulfonate (13)
4.3.5. Synthesis of 4H-pyrrolo [1,2-b]pyrazol-3-yl trifluoromethanesulfonate (14)
4.3.6. Synthesis of 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl trifluoromethanesulfonate (6)
4.4. Synthesis of Withasomnine Homolog 15 (Scheme 3)
4.4.1. Synthesis of 1-allyl-4-iodo-1H-pyrazole (2d)
4.4.2. Synthesis of 5-allyl-1-(but-3-en-1-yl)-4-hydroxy-1H-pyrazole (16)
4.4.3. Synthesis of 5-allyl-1-(but-3-en-1-yl)-1H-pyrazol-4-yl trifluoromethanesulfonate (17)
4.4.4. Synthesis of 7,8-dihydro-4H-pyrazolo[1,5-a]azepin-3-yl trifluoromethanesulfonate (18)
4.4.5. Synthesis of 3-phenyl-7,8-dihydro-4H-pyrazolo[1,5-a]azepine (19)
4.4.6. Synthesis of 3-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a]azepine (15)
4.4.7. Synthesis of 4t from 4b
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. From 2000 to Mid-2010: A fruitful decade for the synthesis of pyrazoles. Chem. Rev. 2011, 111, 6984–7034. [Google Scholar] [CrossRef]
- Ansari, A.; Ali, A.; Asif, M.; Shamsuzzaman, S. Review: Biologically active pyrazole derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-Aizari, F.A.; Ansar, M. Synthesis and pharmacological activities of pyrazole derivatives: A review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.W. Recent developments in the chemistry of pyrazoles. Adv. Heterocycl. Chem. 2018, 126, 55–107. [Google Scholar]
- Usami, Y.; Tatsui, Y.; Yoneyama, H.; Harusawa, S. C4-Alkylamination of C4-halo-1H-1-tritylpyrazoles using Pd(dba)2 or CuI. Molecules 2020, 25, 4634. [Google Scholar] [CrossRef]
- Ray, R.; Hartwig, J.F. Oxalohydrazide ligands for copper-catalyzed C−O coupling reactions with high turnover numbers. Angew. Chem. Int. Ed. 2020, 133, 8284–8292. [Google Scholar] [CrossRef]
- Vijeta, A.; Casadevall, C.; Roy, S.; Reisner, E. Visible-light promoted C-O bond formation with an integrated carbon nitride–nickel heterogeneous photocatalyst. Angew. Chem. Int. Ed. 2021, 60, 8494–8499. [Google Scholar] [CrossRef] [PubMed]
- Vorogushin, A.V.; Huang, X.; Buchwald, S.L. Use of tunable ligands allows for intermolecular Pd-catalyzed C−O Bond formation. J. Am. Chem. Soc. 2005, 127, 8146–8149. [Google Scholar] [CrossRef] [PubMed]
- Maiti, D.; Buchwald, S.L. Orthogonal Cu- and Pd-based catalyst system for the O- and N-arylation of aminophenols. J. Am. Chem. Soc. 2009, 131, 17423–17429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Ruiz-Castillo, P.; Buchwald, S.L. Palladium-catalyzed C-O cross-coupling of primary alcohols. Org. Lett. 2018, 20, 1580–1583. [Google Scholar] [CrossRef] [PubMed]
- Gowrisankar, S.; Sergeev, A.G.; Anbarasan, P.; Spannenberg, A.; Neumann, H.; Beller, M. A general and efficient catalyst for palladium-catalyzed C−O coupling reactions of aryl halides with primary alcohols. J. Am. Chem. Soc. 2010, 132, 11592–11598. [Google Scholar] [CrossRef]
- Maiti, D.; Buchwald, S.L. Cu-catalyzed arylation of phenols: Synthesis of sterically hindered and heteroaryl diaryl ethers. J. Org. Chem. 2010, 75, 1791–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, M.; Batra, S. Copper-Catalyzed Cascade Reactions of Substituted 4-Iodopyrazolecarbaldehydes with 1,2-Phenylenediamines and 2-Aminophenols. Adv. Synth. Catal. 2020, 352, 3431–3437. [Google Scholar] [CrossRef]
- Wong, D.M.; Li, J.; Chen, Q.H.; Han, Q.; Mutunga, J.M.; Wysinski, A.; Anderson, T.D. Select small core structure carbamates exhibit high contact toxicity to “carbamate-resistant” strain malaria mosquitoes, Anopheles gambiae (Akron). PLoS ONE 2012, 7, e46712. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, E.; Ihalainen, E.; Pispa, J.P. Pharmacological and toxicological properties of by 4-hydroxypyrazole, a metabolite of pyrazole. Acta Pharmacol. Toxicol. 1981, 48, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Cornell, N.W.; Hansch, C.; Kim, K.H.; Henegar, K. The inhibition of alcohol dehydrogenase in vitro and in isolated hepatocytes by 4-substituted pyrazoles. Arch. Biochem. Biophys. 1983, 227, 81–90. [Google Scholar] [CrossRef]
- Sinclair, J.; Cornell, N.W.; Zaitlin, L.; Hansch, C. Induction of cytochrome P-450 by alcohols and 4-substituted pyrazoles: Comparison of structure-activity relationships. Biochem. Pharmacol. 1986, 35, 707–710. [Google Scholar] [CrossRef]
- Jones, J.P.; Joswig-Jones, C.A.; Hebner, M.; Chu, Y.; Koop, D.R. The effects of nitrogen-heme-iron coordination on substrate affinities for cytochrome P450 2E1. Chem. Biol. Interact. 2011, 193, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walworth, B.L. Fungicidal Use of 4-alkoxypyrazoles. U.S. (1976) Patent No. US 4000301 A 19761228, 14 November 1975. [Google Scholar]
- Matsuo, J.; Takaya, M.; Maki, Y. 1-Phenyl-4-alkoxypyrazole-5-carboxylic Acid Esters. Jpn. Kokai Tokkyo Koho (1972) Patent No. JP 47042665 A, 16 December 1972. [Google Scholar]
- Munier-Lehmann, H.; Lucas-Hourani, M.; Guillou, S.; Helynck, O.; Zanghi, G.; Noel, A.; Tangy, F.; Vidalain, P.O.; Janin, Y.L. Original 2-(3-Alkoxy-1H-pyrazol-1-yl)pyrimidine derivatives as inhibitors of human dihydroorotate dehydrogenase (DHODH). J. Med. Chem. 2015, 58, 860–877. [Google Scholar] [CrossRef]
- Ichikawa, H.; Watanabe, R.; Fujino, Y.; Usami, Y. Divergent synthesis of withasomnines via synthesis of 4-hydroxy-1H-pyrazoles and Claisen rearrangement of their 4-O-allylethers. Tetrahedron Lett. 2011, 52, 4448–4451. [Google Scholar] [CrossRef]
- Usami, Y.; Watanabe, R.; Fujino, Y.; Shibano, M.; Ishida, C.; Yoneyama, H.; Harusawa, S.; Ichikawa, H. Divergent synthesis and evaluation of inhibitory activities against Cyclooxygenases-1 and −2 of natural withasomnines and analogues. Chem. Pharm. Bull. 2012, 60, 1550–1560. [Google Scholar] [CrossRef] [Green Version]
- Usami, Y.; Tatsui, Y.; Sumimoto, K.; Miyamoto, A.; Koito, N.; Yoneyama, H.; Harusawa, S. 3-Trifluoromethansulfonyloxy-4,7-dihidropyrazolopyridine via ring-closing metathesis: Synthesis and transformation to withasomnine homologs. Heterocycles 2021, 103, 284–299. [Google Scholar] [CrossRef]
- Allin, S.M.; Barton, W.R.S.; Bowman, W.R.; McInally, T. Radical cyclisation onto pyrazoles: Synthesis of withasomnine. Tetrahedron Lett. 2002, 43, 4191–4193. [Google Scholar] [CrossRef]
- Allin, S.M.; Barton, W.R.S.; Russell Bowman, W.; Bridge (née Mann), E.; Elsegood, M.R.J.; McInally, T.; McKee, V. Bu3SnH-mediated radical cyclisation onto azoles. Tetrahedron 2008, 64, 7745–7758. [Google Scholar] [CrossRef]
- Xia, T.; Hu, Z.; Ji, W.; Zhang, S.; Shi, H.; Liu, C.; Pang, B.; Liu, G.; Liao, X. Synthesis of withasomnine and pyrazole derivatives via intramolecular dehydrogenative cyclization, as well as biological evaluation of withasomnine-based scaffolds. Org. Chem. Front. 2018, 5, 850–854. [Google Scholar] [CrossRef]
- Wube, A.A.; Wenzig, E.M.; Gibbons, S.; Asres, K.; Bauer, R.; Bucar, F. Constituents of the stem bark of Discopodium penninervium and their LTB4 and COX-1 and -2 inhibitory activities. PhytoChemistry 2008, 69, 982–987. [Google Scholar] [CrossRef]
- Usami, Y.; Kohno, A.; Yoneyama, H.; Harusawa, S. Synthesis of dihydrooxepino[3,2-c]pyrazoles via Claisen rearrangement and ring-closing metathesis from 4-allyloxy-1H-pyrazoles. Molecules 2018, 23, 592. [Google Scholar] [CrossRef] [Green Version]
- Usami, Y.; Sumimoto, K.; Kishima, A.; Tatsui, Y.; Yoneyama, H.; Harusawa, S. Synthesis of dihydropyrano[3,2-c]pyrazoles via double bond migration and ring-closing metathesis. Molecules 2019, 24, 296. [Google Scholar] [CrossRef] [Green Version]
- Usami, Y.; Tsujiuchi, Y.; Machiya, Y.; Chiba, A.; Ikawa, T.; Yoneyama, H.; Harusawa, S. Synthetic challenges in the construction of 8- to 10-Membered pyrazole-fused rings via ring-closing metathesis. Heterocycles 2020, 101, 496–511. [Google Scholar] [CrossRef]
- Kadoma, Y.; Murakami, Y.; Ogiwara, T.; Machino, M.; Yokoe, I.; Fujisawa, S. Radical-scavenging activity and cytotoxicity of p-methoxyphenol and p-cresol dimers. Molecules 2010, 15, 1103–1112. [Google Scholar] [CrossRef] [Green Version]
- Li, X.B.; Chen, G.Y.; Liu, R.J.; Zheng, C.J.; Song, X.M.; Han, C.R. A new biphenyl derivative from the mangrove endophytic fungus Phomopsis longicolla HL-2232. Nat. Prod. Res. 2017, 31, 2264–2267. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.E.; Walvoord, R.R.; Padilla-Salinas, R.; Kozlowski, M.C. Aerobic copper-catalyzed organic reactions. Chem. Rev. 2013, 113, 6234–6458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, F.; Colombel-Rouen, S.; Dumas, A.; Guégan, J.P.; Roisnel, T.; Dorcet, V.; Baslé, O.; Rouen, M.; Mauduit, M. Activation of olefin metathesis complexes containing unsymmetrical unsaturated N-heterocyclic carbenes by copper and gold transmetalation. Chem. Commun. 2019, 55, 11583–11586. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst | Ligand a | Solvent | Temperature (°C) | Time | 4a Yield, % |
|---|---|---|---|---|---|---|
| 1 b,c | Pd(dba)2 | L1 | xylene | 160 (MW) | 30 min | 0 |
| 2 c | CuI | L2 | DMF | 100 | overnight | 0 |
| 3 c | CuI | L3 | DMF | 100 | overnight | 0 |
| 4 | CuI | L3 | allyl alcohol | 100 | overnight | 51 |
| 5 | CuI | L4 | allyl alcohol | 100 (MW) | 1 h | 31 |
| 6 | CuI | L4 | allyl alcohol | 130 (MW) | 1 h | 66 |
| 7 | CuI | L4 | allyl alcohol | 130 (MW) | 30 min | 24 |
| 8 | CuI | L4 | allyl alcohol | 160 (MW) | 1 h | 16 |
| 9 d | CuI | L4 | allyl alcohol | 130 (MW) | 1 h | 37 |
 . b. 10 mol% Pd(dba)2 was used, c. 2 equiv of allyl alcohol was added, d. 10 mol% CuI was used.
. b. 10 mol% Pd(dba)2 was used, c. 2 equiv of allyl alcohol was added, d. 10 mol% CuI was used.
| Entry | Substrate | R2OH | Product | Yield (%) |
|---|---|---|---|---|
| 1 | 2a: R1 = Tr | R2 = Me | 4c: R1 = Tr, R2 = Me | 61 |
| 2 | 2a | R2 = Et | 4d: R1 = Tr, R2 = Et | 76 |
| 3 | 2a | R2 = n-Pr | 4e: R1 = Tr, R2 = n-Pr | 64 |
| 4 | 2a | R2 = n-Bu | 4f: R1 = Tr, R2 = n-Bu | 33 |
| 5 | 2a | R2 = iPr | 4g: R1 = Tr, R2 = iPr | 9 |
| 6 | 2a | R2 = sec-Bu | 4h: R1 = Tr, R2 = sec-Bu | 0 |
| 7 | 2a | R2 = iBu | 4i: R1 = Tr, R2 = iBu | 45 |
| 8 | 2a | R2 = tert-Bu | 4j: R1 = Tr, R2 = tert-Bu | 0 |
| 9 | 2a | R2 = isoamyl | 4k: R1 = Tr, R2 = isoamyl | 37 |
| 10 a | 2a | R2 = cyclobutyl | 4l: R1 = Tr, R2 = cyclobutyl | 59 |
| 11 a | 2a | R2 = cyclopentyl | 4m: R1 = Tr, R2 = cyclopentyl | 18 |
| 12 a | 2a | R2 = cyclohexyl | 4n: R1 = Tr, R2 = cyclohexyl | 25 |
| 13 b | 2a | R2 = Bn | 4o: R1 = Tr, R2 = Bn | 12 |
| 14 | 2a | R2 = Ph | 4p: R1 = Tr, R2 = Ph | 0 |
| 15 c | 2a | R2 = p-MeOPh | 4q: R1 = Tr, R2 = p-MeOPh | 0 |
| 16 d | 2a | R2 = allyl | 4a: R1 = Tr, R2 = allyl | 66 |
| 17 | 2b: R1 = 1-propenyl | R2 = 1-propenyl | 4r: R1 = 1-propenyl, R2 = allyl | 65 |
| 18 | 2c: R1 = allyl | R2 = allyl | 4s: R1 = R2 = allyl | 58 |
| 19 | 2d: R1 = 3-butenyl | R2 = allyl | 4t: R1 = 3-butenyl, R2 = allyl | 64 |
| 20 e | 2a | R2 = OH | 4u: R1 = Tr, R2 = H | 0 |
| 21 | 1 | R2 = allyl | 4v: R1 = H, R2 = allyl | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usami, Y.; Kubo, Y.; Takagaki, T.; Kuroiwa, N.; Ono, J.; Nishikawa, K.; Nakamizu, A.; Tatsui, Y.; Harusawa, S.; Hayama, N.; et al. CuI-Catalyzed Coupling Reactions of 4-Iodopyrazoles and Alcohols: Application toward Withasomnine and Homologs. Molecules 2021, 26, 3370. https://doi.org/10.3390/molecules26113370
Usami Y, Kubo Y, Takagaki T, Kuroiwa N, Ono J, Nishikawa K, Nakamizu A, Tatsui Y, Harusawa S, Hayama N, et al. CuI-Catalyzed Coupling Reactions of 4-Iodopyrazoles and Alcohols: Application toward Withasomnine and Homologs. Molecules. 2021; 26(11):3370. https://doi.org/10.3390/molecules26113370
Chicago/Turabian StyleUsami, Yoshihide, Yumika Kubo, Toshiki Takagaki, Nao Kuroiwa, Jun Ono, Kohei Nishikawa, Ayaka Nakamizu, Yuya Tatsui, Shinya Harusawa, Noboru Hayama, and et al. 2021. "CuI-Catalyzed Coupling Reactions of 4-Iodopyrazoles and Alcohols: Application toward Withasomnine and Homologs" Molecules 26, no. 11: 3370. https://doi.org/10.3390/molecules26113370
APA StyleUsami, Y., Kubo, Y., Takagaki, T., Kuroiwa, N., Ono, J., Nishikawa, K., Nakamizu, A., Tatsui, Y., Harusawa, S., Hayama, N., & Yoneyama, H. (2021). CuI-Catalyzed Coupling Reactions of 4-Iodopyrazoles and Alcohols: Application toward Withasomnine and Homologs. Molecules, 26(11), 3370. https://doi.org/10.3390/molecules26113370