
Silylated Tag-Assisted Peptide Synthesis: Continuous One-Pot Elongation for the Production of Difficult Peptides under Environmentally Friendly Conditions



Abstract
:
1. Introduction
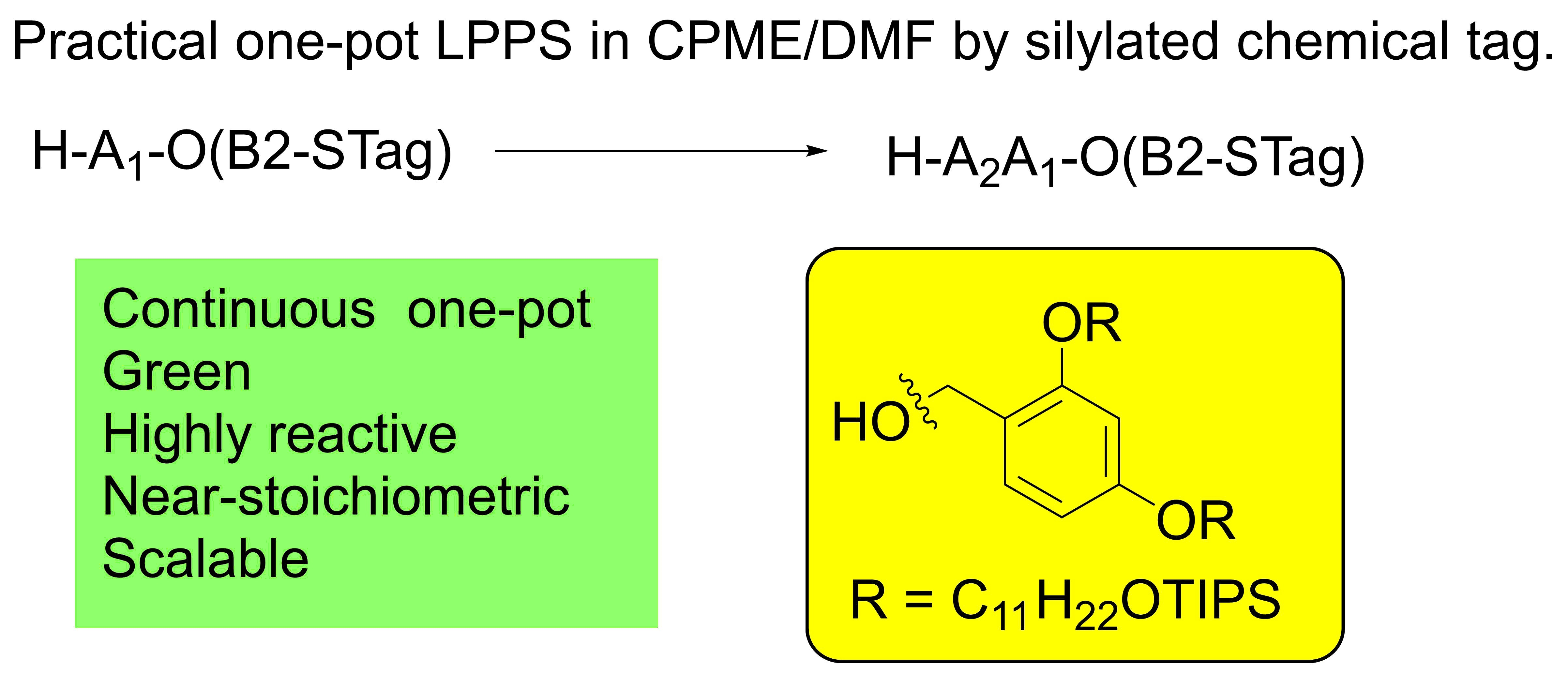
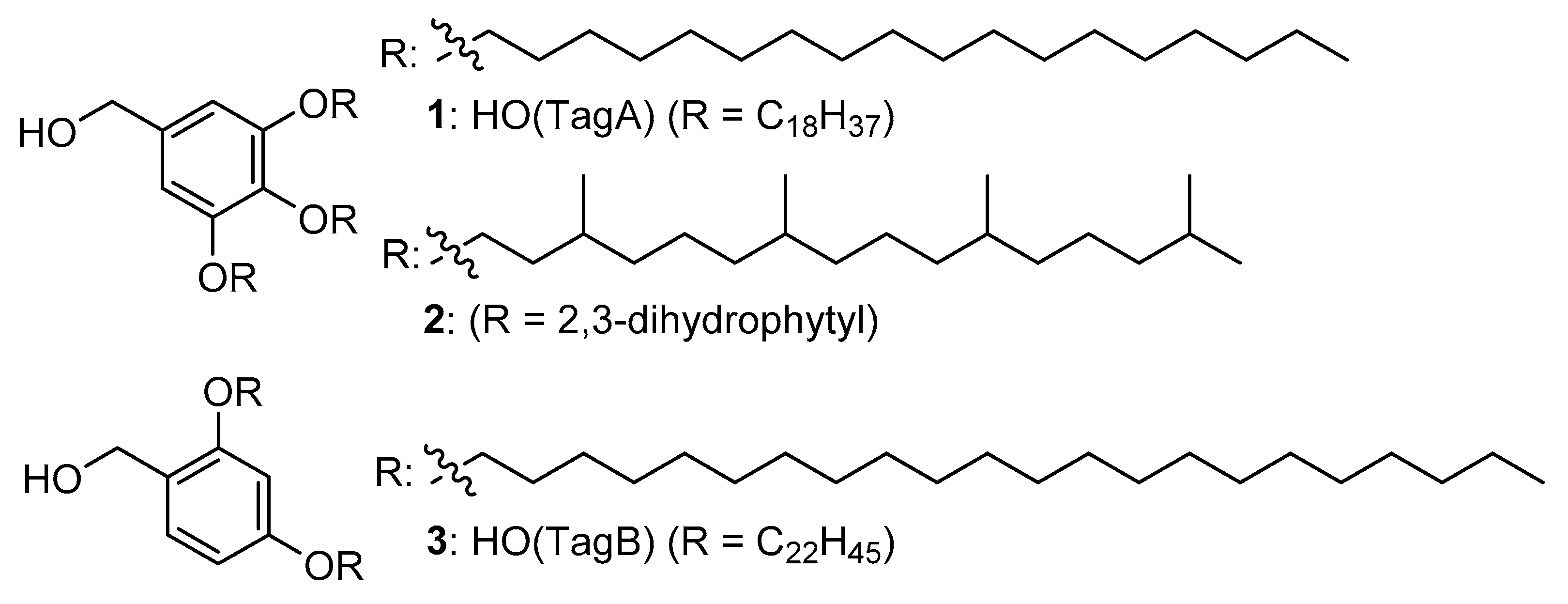
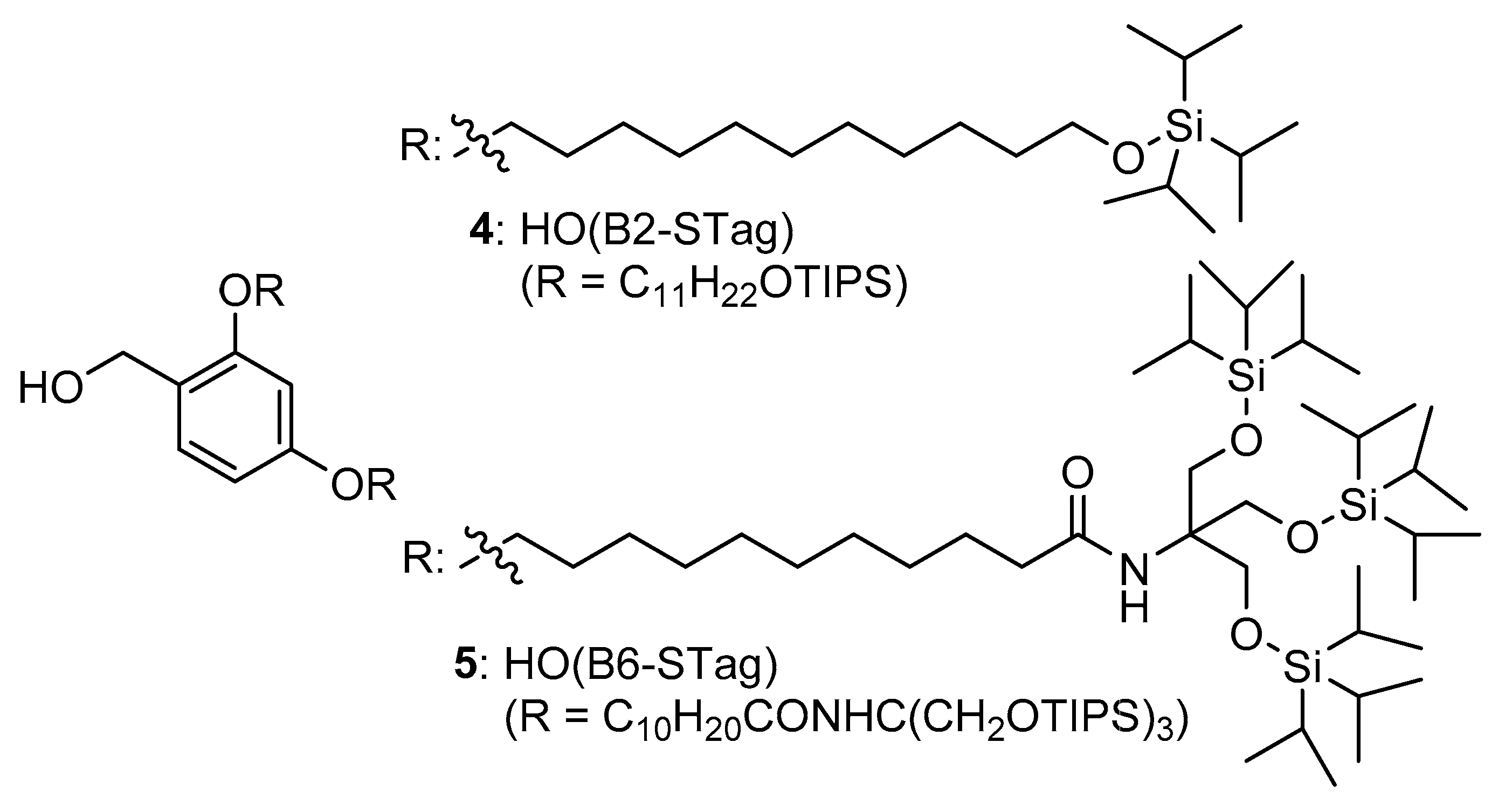
2. Results
3. Discussion
4. Materials and Methods
4.1. Loading: Preparation of H-F-O(B2-STag) (8)
4.2. STag-PS: Synthesis of H-FF-O(B2-STag) (10)
4.3. General Procedure of STag-PS
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Merrifield, R.B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Alshanski, I.; Bentolila, M.; Gitlin-Domagalska, A.; Zamir, D.; Zorsky, S.; Joubran, S.; Hurevich, M.; Gilon, C. Enhancing the efficiency of the solid phase peptide synthesis (SPPS) process by high shear mixing. Org. Process Res. Dev. 2018, 22, 1318–1322. [Google Scholar] [CrossRef]
- Hwang, T.-L.; Ranganathan, K.; Fang, Y.-Q.; Crockett, R.D.; Osgood, S.; Cui, S. Studies of Coupling Kinetics and Correlation of Reaction Conversions to Color Tests for Solid-Phase Peptide Synthesis of AMG 416 by NMR. Org. Process Res. Dev. 2018, 22, 1007–1014. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Kenworthy, M.N.; Mukherjee, S.; Kopach, M.E.; Wegner, K.; Gallou, F.; Smith, A.G.; Roschangar, F. Sustainability challenges in peptide synthesis and purification: From R&D to production. J. Org. Chem. 2019, 84, 4615–4628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamiaki, H.; Obata, T.; Azefu, Y.; Toma, K. A novel protecting group for constructing combinatorial peptide libraries. Bull. Chem. Soc. Jpn. 2001, 74, 733–738. [Google Scholar] [CrossRef]
- Okada, Y.; Suzuki, H.; Nakae, T.; Fujita, S.; Abe, H.; Nagano, K.; Yamada, T.; Ebata, N.; Kim, S.; Chiba, K. Tag-assisted liquid-phase peptide synthesis using hydrophobic benzyl alcohols as supports. J. Org. Chem. 2013, 78, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Yano, T.; Fukui, T. Novel diphenylmethyl-derived amide protecting group for efficient liquid-phase peptide synthesis: AJIPHASE. Org. Lett. 2012, 14, 4514–4517. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Inomata, T.; Fukui, T. AJIPHASE®: A Highly Efficient Synthetic Method for One-Pot Peptide Elongation in the Solution Phase by an Fmoc Strategy. Angew. Chem., Int. Ed. 2017, 56, 7803–7807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, S.; Boughtflower, B.; Mutton, I.; Paterson, C.; Farrant, D.; Taylor, N.; Blaxill, Z.; Carmody, C.; Borman, P. Toward single-calibrant quantification in HPLC. A comparison of three detection strategies: Evaporative light scattering, chemiluminescent nitrogen, and proton NMR. Anal. Chem. 2005, 77, 4354–4365. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, D.S.; Murray, J.; Sneddon, H.F.; Jamiesona, C.; Watson, A.J.B. Evaluation of alternative solvents in common amide coupling reactions: Replacement of dichloromethane and N, N-dimethylformamide. Green Chem. 2013, 15, 596–600. [Google Scholar] [CrossRef] [Green Version]
- Carpino, L.A.; Ghassemi, S.; Ionescu, D.; Ismail, M.; Sadat-Aalaee, D.; Truran, G.A.; Mansour, E.M.E.; Siwruk, G.A.; Eynon, J.S.; Morgan, B. Rapid, continuous solution-phase peptide synthesis: Application to peptides of pharmaceutical interest. Org. Process Res. Dev. 2003, 7, 28–37. [Google Scholar] [CrossRef]
- Chiba, K.; Kono, Y.; Kim, S.; Nishimoto, K.; Kitanoa, Y.; Tada, M. A liquid-phase peptide synthesis in cyclohexane-based biphasic thermomorphic systems. Chem. Commun. 2002, 1766–1767. [Google Scholar] [CrossRef]
- Watanabe, K.; Yamagiwa, N.; Torisawa, Y. Cyclopentyl methyl ether as a new and alternative process solvent. Org. Process Res. Dev. 2007, 11, 251–258. [Google Scholar] [CrossRef]
- Subirós-Funosas, R.; Nieto-Rodriguez, L.; Jensen, K.J.; Albericio, F. COMU: Scope and limitations of the latest innovation in peptide acyl transfer reagents. J. Pept. Sci. 2013, 19, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.A.; Yin, Y.; Suga, H. Macrocyclic peptides as drug candidates: Recent progress and remaining challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.S.; Shepherd, N.E.; Xu, W.; Lucke, A.J.; Stoermer, M.J.; Fairlie, D.P. Orally absorbed cyclic peptides. Chem. Rev. 2017, 117, 8094–8128. [Google Scholar] [CrossRef] [PubMed]
- Falb, E.; Yechezkel, T.; Salitra, Y.; Gilon, C. In situ generation of Fmoc-amino acid chlorides using bis-(trichloromethyl) carbonate and its utilization for difficult couplings in solid-phase peptide synthesis. J. Peptide Res. 1999, 53, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-W.; Grossmann, T.N.; Verdine, G.L. Synthesis of all-hydrocarbon stapled α-helical peptides by ring-closing olefin metathesis. Nat. Protoc. 2011, 6, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Jastrzabek, K.G.; Subiros-Funosas, R.; Albericio, F.; Kolesinska, B.; Kaminski, Z.J. 4-(4, 6-Di [2, 2, 2-trifluoroethoxy]-1, 3, 5-triazin-2-yl)-4-methylomorpholinium Tetrafluoroborate. Triazine-Based Coupling Reagents Designed for Coupling Sterically Hindered Substrates. J. Org. Chem. 2011, 76, 4506–4513. [Google Scholar] [CrossRef] [PubMed]
- For the Concept of Greener Chemical Process or Product. Available online: https://www.acs.org/content/acs/en/greenchemistry/principles/12-principles-of-green-chemistry.html (accessed on 7 June 2021).
- Kumar, A.; Jad, Y.E.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Green solid-phase peptide synthesis 4. γ-Valerolactone and N-formylmorpholine as green solvents for solid phase peptide synthesis. Tetrahedron Lett. 2017, 58, 2986–2988. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide a | Solubility (mM, 25 °C) | ||||
|---|---|---|---|---|---|
| CPME | CPME/DMF (7/3) | DMF | THF | ||
| entry 1 | Fmoc-FLG-O(TagA) b | 3 | 46 | 6 | 32 |
| entry 2 | Fmoc-FLG-O(TagB) c | 4 | 35 | - d | 45 |
| entry 3 | Fmoc-FLG-O(B2-STag) | 124 | 341 | 509 | 238 |
| entry 4 | Fmoc-FLG-O(B6-STag) | 549 | 790 | >1910 | - |
| entry 5 | H-FLG-O(TagA) | 105 | 378 | 51 | - |
| entry 6 | H-FLG-O(TagB) | 112 | 113 | - | 193 |
| entry 7 | H-FLG-O(B2-STag) | >1630 | >2230 | >2420 | - |
| entry 8 | H-FLG-O(B6-STag) | >1930 | >2320 | >2430 | - |

| Solvent | Conc. of (6) (mM) | Conversion (%) a | |||
|---|---|---|---|---|---|
| 10 Min | 30 Min | 60 Min | |||
| entry 1 | CPME/DMF (7/3) | 10 | 43 | 77 | 91 |
| entry 2 | 100 | 94 | 99 | 99.3 | |
| entry 3 | DMF | 10 | 27 | 56 | 74 |
| entry 4 | 100 | 81 | 95 | 98 | |
| entry 7 | CPME | 10 | 0 | 1 | 3 |
| entry 8 | 100 | 1 | 5 | 13 | |

| Additive; Quenching | H-FFFF-OH (%) b | H-FFFFF-OH (%) b | |
|---|---|---|---|
| entry 1 | Oxyma; none | 96.8 | 2.8 |
| entry 2 | none; none | 99.4 | 0.4 |
| entry 3 | Oxyma; AEE | 99.6 | 0.3 |
| entry 4 | Oxyma; AEHS | 99.7 | 0.1 |

| Peptide | Purity (%) b | Des-X (%) b | Des-YX (%) b | |
|---|---|---|---|---|
| entry 1 | H-YAAFL-OH | 98.5 | n.d. c | n.d. |
| entry 2 | H-Y(MeL)(MeL)FL-OH | 96.6 | 0.4 | 0.6 |
| entry 3 | H-Y(MeF)(MeF)FL-OH | 91.5 | 2.7 | 0.5 |
| General Procedure | Loading | |
|---|---|---|
| substrate | STagged peptide: 1.26 mmol | STag (4): 1.26 mmol |
| Fmoc-amino acid | 1.51 mmol | 3.78 mmol |
| coupling reagent | COMU: 1.51 mmol | EDCI: 3.78 mmol |
| base | DIPEA: 5.04 mmol | DMAP a: 0.13 mmol |
| AEE | 0.76 mmol | 3.02 mmol |
| MPSNa | 3.02 mmol | 11.34 mmol |
| DMSO | 2.5 mL | 9.5 mL |
| DBU | 6.55 mmol | 16.4 mmol |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yano, S.; Mori, T.; Kubota, H. Silylated Tag-Assisted Peptide Synthesis: Continuous One-Pot Elongation for the Production of Difficult Peptides under Environmentally Friendly Conditions. Molecules 2021, 26, 3497. https://doi.org/10.3390/molecules26123497
Yano S, Mori T, Kubota H. Silylated Tag-Assisted Peptide Synthesis: Continuous One-Pot Elongation for the Production of Difficult Peptides under Environmentally Friendly Conditions. Molecules. 2021; 26(12):3497. https://doi.org/10.3390/molecules26123497
Chicago/Turabian StyleYano, Shinya, Toshihiro Mori, and Hideki Kubota. 2021. "Silylated Tag-Assisted Peptide Synthesis: Continuous One-Pot Elongation for the Production of Difficult Peptides under Environmentally Friendly Conditions" Molecules 26, no. 12: 3497. https://doi.org/10.3390/molecules26123497
APA StyleYano, S., Mori, T., & Kubota, H. (2021). Silylated Tag-Assisted Peptide Synthesis: Continuous One-Pot Elongation for the Production of Difficult Peptides under Environmentally Friendly Conditions. Molecules, 26(12), 3497. https://doi.org/10.3390/molecules26123497