
Benzylated Dihydroflavones and Isoquinoline-Derived Alkaloids from the Bark of Diclinanona calycina (Annonaceae) and Their Cytotoxicities

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
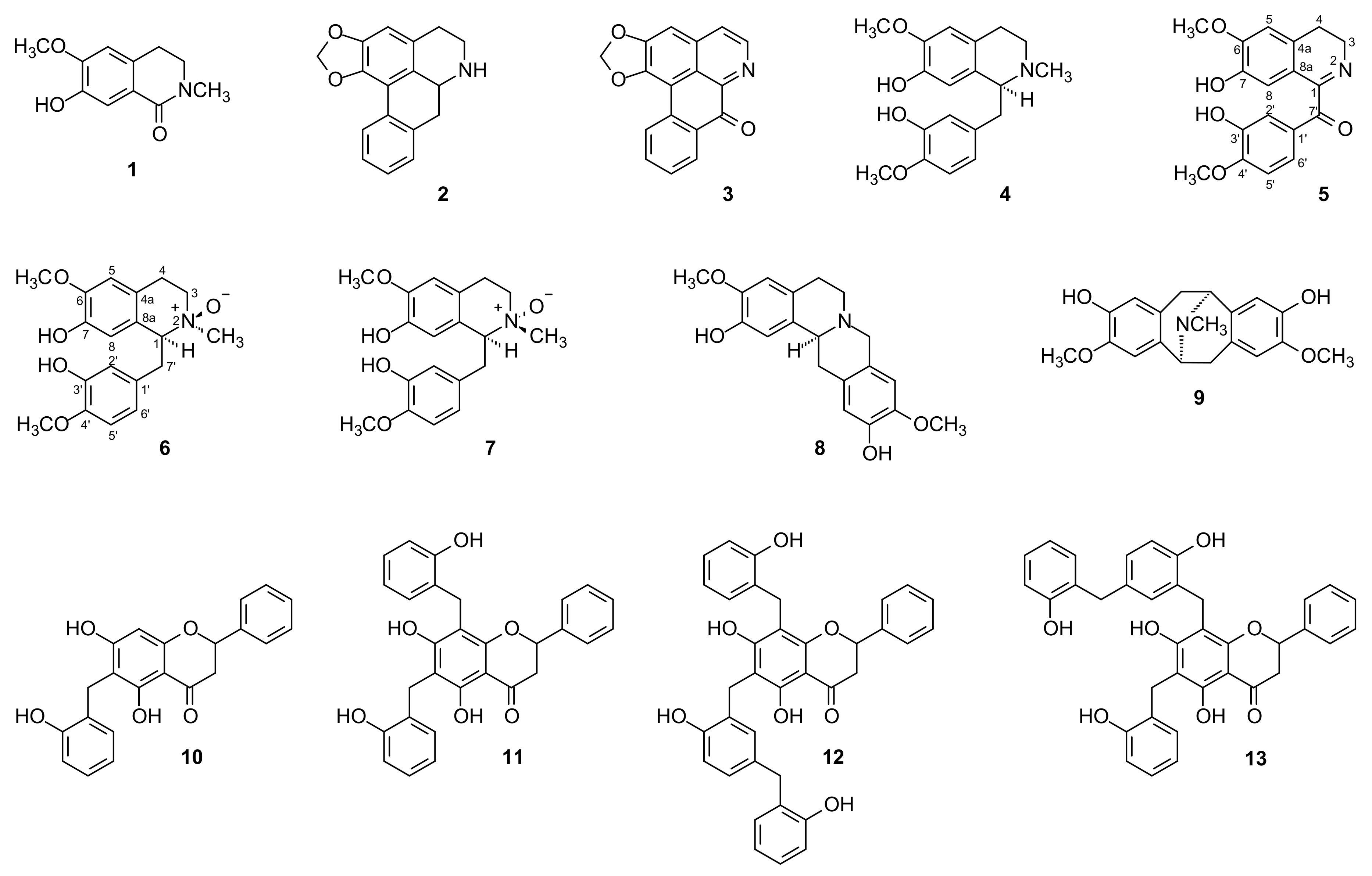
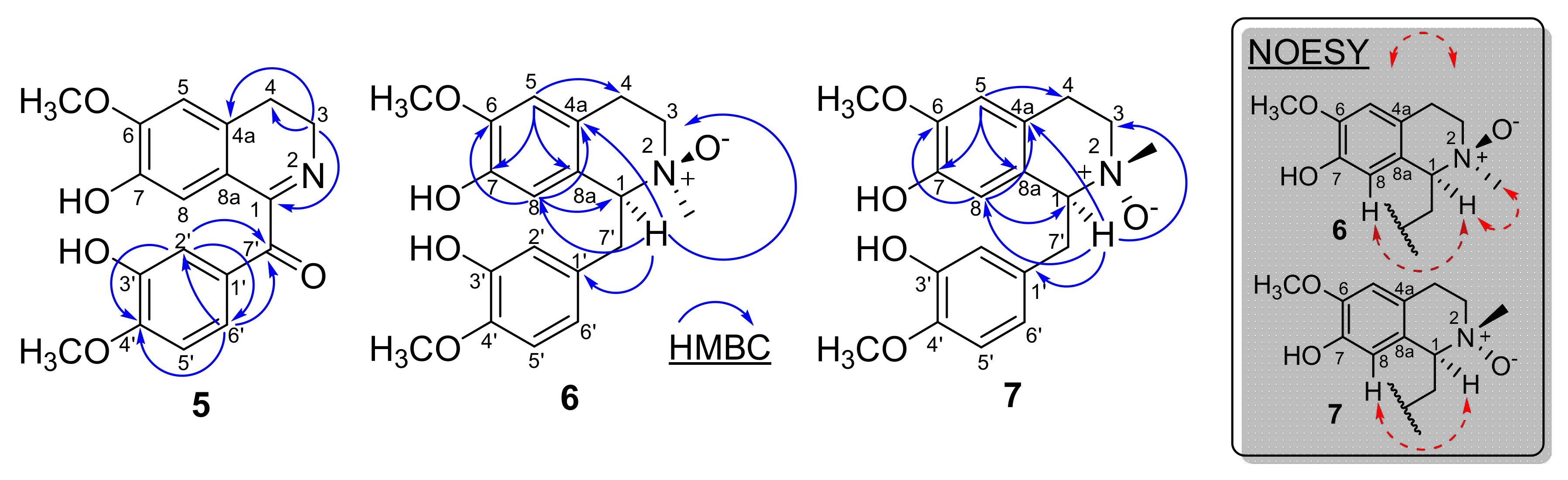
2.1. Structural Elucidation of the Compounds
2.2. Cytotoxic Assay
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. In Vitro Cytotoxic Assay
3.4.1. Cells
3.4.2. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Couvreur, T.L.P.; Pirie, M.D.; Chatrou, L.W.; Saunders, R.M.K.; Su, Y.C.F.; Richardson, J.E.; Erkens, R.H.J. Early evolutionary history of the flowering plant family Annonaceae: Steady diversification and boreotropical geodispersal. J. Biogeogr. 2011, 38, 664–680. [Google Scholar] [CrossRef]
- Corrêa, M.P. Dicionário das Plantas Úteis do Brasil e das Exóticas Cultivadas; IBDF Ministério da Agricultura: Rio de Janeiro, Brazil, 1984. [Google Scholar]
- Leboeuf, M.; Cave, A.; Bhaumik, P.K.; Mukherjee, B.; Mukherjee, R. The phytochemistry of the Annonaceae. Phytochemistry 1982, 21, 2783–2813. [Google Scholar] [CrossRef]
- Rupprecht, J.K.; Hui, Y.-H.; McLaughlin, J.L. Annonaceous acetogenins: A review. J. Nat. Prod. 1990, 53, 237–278. [Google Scholar] [CrossRef]
- Lúcio, A.S.S.C.; Almeida, J.R.G.D.S.; Da-Cunha, E.V.L.; Tavares, J.F.; Filho, J.M.B. Alkaloids of the Annonaceae: Occurrence and a compilation of their biological activities. In The Alkaloids; Knolker, H.-J., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; Chapter 5; Volume 74, pp. 233–409. [Google Scholar] [CrossRef]
- Souza, C.A.S.; Nardelli, V.B.; Paz, W.H.P.; Pinheiro, M.L.B.; Rodrigues, A.C.B.C.; Bomfim, L.M.; Soares, M.B.P.; Bezerra, D.P.; Chaar, J.S.; Koolen, H.H.F.; et al. Asarone-derived phenylpropanoids and isoquinoline-derived alkaloids from the bark of Duguetia pycnastera (Annonaceae) and their cytotoxicities. Quim. Nova 2020, 43, 1397–1403. [Google Scholar] [CrossRef]
- Araújo, M.S.; Silva, F.M.A.; Koolen, H.H.F.; Costa, E.V. Isoquinoline-derived alkaloids from the bark of Guatteria olivacea (Annonaceae). Biochem. Syst. Ecol. 2020, 92, 104105. [Google Scholar] [CrossRef]
- Ngouonpe, A.W.; Mbobda, A.S.W.; Happi, G.M.; Mbiantcha, M.; Tatuedom, O.K.; Ali, M.S.; Lateef, M.; Tchouankeu, J.C.; Kouam, S.F. Natural products from the medicinal plant Duguetia staudtii (Annonaceae). Biochem. Syst. Ecol. 2019, 83, 22–25. [Google Scholar] [CrossRef]
- Santos, R.C.; Souza, A.V.; Andrade-Silva, M.; Cruz, K.C.V.; Kassuya, C.A.L.; Cardoso, C.A.L.; Vieira, M.C.; Formagio, A.S.N. Antioxidant, anti-rheumatic and anti-inflammatory investigation of extract and dicentrinone from Duguetia furfuracea (A. St.-Hil.) Benth. & Hook. f. J. Ethnopharmacol. 2018, 211, 9–16. [Google Scholar] [CrossRef]
- Costa, E.V.; Pinheiro, M.L.B.; Souza, A.D.L.; Barison, A.; Campos, F.R.; Valdez, R.H.; Ueda-Nakamura, T.; Dias Filho, B.P.; Nakamura, C.V. Trypanocidal activity of oxoaporphine and pyrimidine-β-carboline alkaloids from the branches of Annona foetida Mart. (Annonaceae). Molecules 2011, 16, 9714–9720. [Google Scholar] [CrossRef]
- Silva, D.B.; Tulli, E.C.O.; Milita, G.C.G.; Costa-Lotufo, L.V.; Pessoa, C.; Moraes, M.O.; Albuquerque, S.; Siqueira, J.M. The antitumoral, trypanocidal and antileishmanial activities of extract and alkaloids isolated from Duguetia furfuracea. Phytomedicine 2009, 16, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.V.; Pinheiro, M.L.B.; Xavier, C.M.; Silva, J.R.A.; Amaral, A.C.F.; Souza, A.D.L.; Barison, A.; Campos, F.R.; Ferreira, A.G.; Machado, G.M.C.; et al. A pyrimidine-β-carboline and other alkaloids from Annona foetida with antileishmanial activity. J. Nat. Prod. 2006, 69, 292–294. [Google Scholar] [CrossRef]
- Pérez, E.; Sáez, J.; Blair, S.; Franck, X.; Figadère, B. Isoquinoline alkaloids from Duguetia vallicola stem bark with antiplasmodial activity. Lett. Org. Chem. 2004, 1, 102–104. [Google Scholar] [CrossRef]
- Costa, E.V.; Teixeira, S.D.; Marques, F.A.; Duarte, M.C.T.; Delarmelina, C. Chemical composition and antimicrobial activity of the essential oils of the Amazon Guatteriopsis species. Phytochemistry 2008, 69, 1895–1899. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.V.; Cruz, P.E.O.; Lourenço, C.C.; Moraes, V.R.S.; Nogueira, P.C.L.; Salvador, M.J. Antioxidant and antimicrobial activities of aporphinoids and other alkaloids from the bark of Annona salzmannii A. DC. (Annonaceae). Nat. Prod. Res. 2013, 27, 1002–1006. [Google Scholar] [CrossRef]
- Costa, R.G.A.; Anunciação, T.A.; Araújo, M.S.; Souza, C.A.; Rosane, B.D.; Sales, C.B.S.; Rocha, C.A.G.; Soares, M.B.P.; Silva, F.M.A.; Koolen, H.H.F.; et al. In vitro and in vivo growth inhibition of human acute promyelocytic leukemia HL-60 cells by Guatteria megalophylla Diels (Annonaceae) leaf essential oil. Biomed. Pharmacother. 2020, 122, 109713. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.V.; Sampaio, M.F.C.; Menezes, L.R.A.; Dutra, L.M.; Costa, C.O.S.; Pinheiro, M.L.B.; Silva, F.M.A.; Soares, M.B.P.; Bezerra, P.B.; Barison, A.; et al. Diversity of the diterpenes in the leaves of Xylopia laevigata (Annonaceae) and their cytotoxicities. Quim. Nova 2020, 43, 419–425. [Google Scholar] [CrossRef]
- Paz, W.H.P.; Oliveira, R.N.; Heerdt, G.; Angolini, C.F.F.; Medeiros, L.S.; Silva, V.R.; Santos, L.S.; Soares, M.B.P.; Bezerra, D.P.; Morgon, N.H.; et al. Structure-based molecular networking for the target discovery of oxahomoaporphine and 8-oxohomoaporphine alkaloids from Duguetia surinamensis. J. Nat. Prod. 2019, 82, 2220–2228. [Google Scholar] [CrossRef]
- Jacobo-Herrera, N.; Pérez-Plasencia, C.; Castro-Torres, V.A.; Martínez-Vázquez, M.; González, A.R. Selective acetogenins and their potential as anticancer agents. Front. Pharmacol. 2020, 10, 783. [Google Scholar] [CrossRef]
- Menezes, L.R.A.; Costa, C.O.D.; Rodrigues, A.C.B.C.; Santo, F.R.E.; Nepel, A.; Dutra, L.M.; Silva, F.M.A.; Soares, M.B.P.; Barison, A.; Costa, E.V.; et al. Cytotoxic Alkaloids from the Stem of Xylopia laevigata. Molecules 2016, 21, 890. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.A.; Barros, G.A.; Silva, J.N.; Oliveira, K.M.; Bezerra, D.P.; Soares, M.B.P.; Costa, E.V. Experimental and theoretical study on spectral features, reactivity, solvation, topoisomerase I inhibition and in vitro cytotoxicity in human HepG2 cells of guadiscine and guadiscidine aporphine alkaloids. J. Mol. Struct. 2021, 1229, 129844. [Google Scholar] [CrossRef]
- Chatrou, L.W.; Pirie, M.D.; Erkens, R.H.J.; Couvreur, T.L.P.; Neubig, K.M.; Abbott, J.R.; Mols, J.B.; Maas, J.W.; Saunders, R.M.K.; Chase, M.W. A new subfamilial and tribal classification of the pantropical flowering plant family Annonaceae informed by molecular phylogenetics. Bot. J. Linn. Soc. 2012, 169, 5–40. [Google Scholar] [CrossRef] [Green Version]
- Maas, P.J.M.; Maas, H.; Miralha, J.M.S.; Junikka, L. Flora da Reserva Ducke, Amazonas, Brasil: Annonaceae. Rodriguésia 2007, 58, 617–662. [Google Scholar] [CrossRef] [Green Version]
- Erkens, R.H.J.; Chatrou, L.W.; Chaowasku, T.; Westra, L.Y.T.; Maas, J.W.; Maas, P.J.M. A decade of uncertainty: Resolving the phylogenetic position of Diclinanona (Annonaceae), including taxonomic notes and a key to the species. Taxon 2014, 63, 1244–1252. [Google Scholar] [CrossRef]
- Carneiro, A.L.B.; Teixeira, M.F.S.; Oliveira, V.M.A.; Fernandes, O.C.C.; Cauper, G.S.B.; Pohlit, A.M. Screening of Amazonian plants from the Adolpho Ducke forest reserve, Manaus, state of Amazonas, Brazil, for antimicrobial activity. Mem. Inst. Oswaldo Cruz 2008, 103, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castilho, A.L.; Silva, J.P.C.; Saraceni, C.H.C.; Díaz, I.E.C.; Paciencia, M.L.B.; Varella, A.D.; Suffredinil, I.B. In vitro activity of Amazon plant extracts against Enterococcus faecalis. Braz. J. Microbiol. 2014, 45, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Cruz, P.E.O.; Costa, E.V.; Moraes, V.R.S.; Nogueira, P.C.L.; Vendramin, M.E.; Andersson, B.; Ferreira, A.G.; Prata, A.P.N. Chemical constituents from the bark of Annona salzmannii (Annonaceae). Biochem. Syst. Ecol. 2011, 39, 872. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-S.; Lai, Y.-C.; Chen, C.-K.; Tseng, L.-H.; Wang, C.-Y. Characterization of isoquinoline alkaloids from Neolitsea sericea var. aurata by HPLC-SPE-NMR. J. Nat. Prod. 2007, 70, 637–642. [Google Scholar] [CrossRef]
- Zhang, M.-S.; Yang, F.-M.; Wang, D.-P.; Zhang, J.-X.; Sun, Q.-Y.; Liang, G.-Y.; Pan, W.-D. Novel N-oxide derivatives of benzyltetrahydroisoquinoline alkaloid from the tubers of Stephania viridiflavens H.S. Lo et M. Yang. Phytochem. Lett. 2012, 5, 96–99. [Google Scholar] [CrossRef]
- Zhou, Q.; Fu, Y.-H.; Li, X.-B.; Chen, G.-Y.; Wu, S.-Y.; Song, X.-P.; Liu, Y.-P.; Han, C.-R. Bioactive benzylisoquinoline alkaloids from Artabotrys hexapetalus. Phytochem. Lett. 2015, 11, 296–300. [Google Scholar] [CrossRef]
- Dörnyei, G.; Bárczai-Beke, M.; Blaskó, G.; Péchy, P.; Szántay, C. Studies aimed at the synthesis of morphine V an economic approach to (±)-reticuline from 3,4-dihydropapaverine. Tetrahedron Lett. 1982, 23, 2913–2916. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, B.W.; Kang, N.S.; Moon, Y.H.; Yang, M.S.; Park, K.H. Alkaloids from the stem bark of Phellodendron amurense Rupr. J. Life Sci. 2005, 15, 423–426. [Google Scholar] [CrossRef]
- Costa, E.V.; Sampaio, M.F.C.; Salvador, M.S.; Nepel, A.; Barison, A. Chemical constituents from the stem of Annona pickelii (Annonaceae). Quim. Nova 2015, 38, 769–776. [Google Scholar] [CrossRef]
- Costa, E.V.; Marques, F.A.; Pinheiro, M.L.B.; Vaz, N.P.; Duarte, M.C.T.; Delarmelina, C.; Braga, R.M.; Maia, B.H.L.N.S. 7,7-Dimethylaporphine alkaloids from the stem of Guatteriopsis friesiana. J. Nat. Prod. 2009, 72, 1516–1519. [Google Scholar] [CrossRef]
- Costa, E.V. Estudo Fitoquímico e Atividades Biológicas de Guatteriopsis blepharophylla, Guatteriopsis friesiana e Guatteriopsis hispida (Annonaceae). Ph.D. Thesis, Doutorado em Química. Programa de Pós-Graduação em Química, Universidade Federal do Paraná, Curitiba, Brazil, 2009. [Google Scholar]
- Stadler, R.; Kutchan, T.M.; Zenk, M.H. (S)-Norcoclaurine is the central intermediate in benzylisoquinoline alkaloid biosynthesis. Phytochemistry 1989, 28, 1083–1086. [Google Scholar] [CrossRef]
- Achenbach, H.; Höhn, M.; Waibel, R.; Nkunya, M.H.H.; Jonker, A.; Muhie, S. Oxygenated pyrenes, their potential biosynthetic precursor and benzylated dihydroflavones from two African Uvaria species. Phytochemistry 1997, 44, 359–364. [Google Scholar] [CrossRef]
- Panichpol, K.; Waterman, P.G. Novel flavonoids from the stem of Popowia caufiflora. Phytochemistry 1978, 17, 1363–1367. [Google Scholar] [CrossRef]
- Lasswell, W.L.; Hufford, C.D. Cytotoxic C-benzylated flavonoids from Uvaria chamae. J. Org. Chem. 1977, 42, 1295–1302. [Google Scholar] [CrossRef]
- Hufford, C.D.; Lasswell, W.L.; Hirotsu, K.; Clardy, J. Uvarinol: A novel cytotoxic tribenzylated flavanone from Uvaria chamae. J. Org. Chem. 1979, 44, 4709–4710. [Google Scholar] [CrossRef]
- Hufford, C.D.; Lasswell, W.L. Uvaretin and isouvaretin. Two novel cytotoxic C-benzylflavanones from Uvaria chamae L. J. Org. Chem. 1976, 41, 1297–1298. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Torrance, S.J.; Wiedhopf, R.M.; Arora, S.K.; Bates, R.B. Uvaretin, a new antitumor agent from Uvaria acuminata (Annonaceae). J. Org. Chem. 1976, 41, 1852–1855. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.V.; Pinheiro, M.L.B.; Maia, B.H.L.N.S.; Marques, F.A.; Ruiz, A.L.T.G.; Marchetti, G.M.; Carvalho, J.E.; Soares, M.B.P.; Costa, C.O.S.; Galvão, A.F.C.; et al. 7,7-Dimethylaporphine and other alkaloids from the bark of Guatteria friesiana. J. Nat. Prod. 2016, 79, 1524–1531. [Google Scholar] [CrossRef] [PubMed]
- Zidorn, C. Plant chemophenetics—A new term for plant chemosystematics/plant chemotaxonomy in the macro-molecular era. Phytochemistry 2019, 163, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Kamada, T.; Phan, C.-S.; Sien, V.S.-T.; Vairappan, C.S. Halogenated chamigrane sesquiterpenes from Bornean Laurencia majuscula. J. Appl. Phycol. 2018, 30, 3373–3378. [Google Scholar] [CrossRef]
- Jensen, S.R.; Franzyk, H.; Wallander, E. Chemotaxonomy of the oleaceae: Iridoids as taxonomic markers. Phytochemistry 2002, 60, 213–231. [Google Scholar] [CrossRef]
- Ahmed, S.A.; Gogal, R.M., Jr.; Walsh, J.E. A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: An alternative to [3H]-thymidine incorporation assay. J. Immunol. Methods 1994, 170, 211–224. [Google Scholar] [CrossRef]
- Santos, L.S.; Silva, V.R.; Menezes, L.R.A.; Soares, M.B.P.; Costa, E.V.; Bezerra, D.P. Xylopine induces oxidative stress and causes G2/M phase arrest, triggering caspasemediated apoptosis by p53-independent pathway in HCT116 cells. Oxid. Med. Cell. Longev. 2017, 2017, 7126872. [Google Scholar] [CrossRef] [Green Version]
- Silva, V.R.; Corrêa, R.S.; Santos, L.S.; Soares, M.B.P.; Batista, A.A.; Bezerra, D.P. A ruthenium-based 5-fluorouracil complex with enhanced cytotoxicity and apoptosis induction action in HCT116 cells. Sci. Rep. 2018, 8, 288. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Position | 5 | 6 | 6 e | 7 | 7 e | |||
|---|---|---|---|---|---|---|---|---|
| δC Mult. a,c,d | δH Mult. a (J in Hz) | δC Mult. b,c,d | δH Mult. b (J in Hz) | δH Mult. | δC Mult. b,c,d | δH Mult. b (J in Hz) | δH Mult. | |
| 1 | 165.0 | 79.8 | 4.13 br d (J 10.4) | 4.82 br d | 79.4 | 4.46 dd (J 8.7; 2.9) | 5.08 dd | |
| 3α 3β | 47.2 | 3.89 t (J 7.8) | 60.7 | 3.42 m 3.76 m | N.D. | 62.9 | 3.48 m 3.67 m | N.D. |
| 4α 4β | 25.4 | 2.79 t (J 7.8) | 27.0 | 3.14 m | N.D. | 26.3 | 2.93 dt (J 17.2; 5.8) 3.21 dt (J 17.2; 7.3) | N.D. |
| 4a | 130.1 | 120.6 | 122.8 | |||||
| 5 | 109.9 | 6.71 s | 112.5 | 6.76 s | 6.79 s | 112.3 | 6.71 s | 6.77 s |
| 6 | 149.0 | 149.4 | 148.9 | |||||
| 7 | 144.2 | 145.7 | 146.1 | |||||
| 8 | 113.0 | 6.90 s | 115.7 | 5.78 s | 5.75 s | 115.5 | 6.30 s | 6.05 s |
| 8a | 120.1 | 127.1 | 126.9 | |||||
| 1′ | 129.1 | 131.2 | 131.4 | |||||
| 2′ | 116.0 | 7.57 d (J 2.0) | 118.0 | 6.58 d (J 2.2) | 6.54 d | 117.3 | 6.65 d (J 2.0) | 6.63 d |
| 3′ | 145.4 | 147.6 | 147.9 | |||||
| 4′ | 151.4 | 148.0 | 148.2 | |||||
| 5′ | 109.9 | 6.88 d (J 8.4) | 112.8 | 6.81 d (J 8.2) | 6.82 d | 113.1 | 6.83 d (J 8.2) | 6.85 d |
| 6′ | 124.3 | 7.60 dd (J 8.4; 2.0) | 122.5 | 6.47 dd (J 8.2; 2.2) | 6.48 br d | 121.6 | 6.60 dd (J 8.2; 2.0) | 6.56 br d |
| 7′α 7′β | 192.8 | 38.9 | 4.04 dd (J 12.6; 2.5) 2.54 dd (J 12.6; 10.4) | N.D. | 39.0 | 3.62 dd (J 14.2; 2.9) 2.79 dd (J 14.2; 8.7) | N.D. | |
| H3CO-6 | 56.0 | 3.93 s | 56.4 | 3.81 s | 3.82 | 56.5 | 3.80 s | 3.82 s |
| H3CO-4′ | 56.1 | 3.95 s | 56.4 | 3.82 s | 3.81 | 56.4 | 3.81 s | 3.81 s |
| H3C-NO | 56.3 | 3.15 s | 3.46 | 54.6 | 3.20 s | 3.59 s | ||
| Compounds | IC50 in µg·mL−1 (µmol·L−1) a | ||||
|---|---|---|---|---|---|
| HL-60 | MCF-7 | HepG2 | HCT116 | MRC-5 | |
| Thalifoline (1) | N.D | N.D | 20.08 (96.96) 17.15−23.51 | >25.0 (>120.72) | >25.0 (>120.72) |
| (S)-(+)-Reticuline (4) | N.D | N.D | 22.54 (68.47) 17.39−29.21 | >25.0 (>75.95) | >25.0 (>75.95) |
| 1S,2R-Reticuline Nβ-oxide (6) | N.D | N.D | 23.11 (66.95) 19.50−36.22 | >25.0 (>72.43) | >25.0 (>72.43) |
| 1S,2S-Reticuline Nα-oxide (7) | N.D | N.D | >25.0 (>72.43) | >25.0 (>72.43) | >25.0 (>72.43) |
| Bisnorargemonine (9) | N.D | N.D | >25.0 (>72.43) | >25.0 (>72.43) | >25.0 (>72.43) |
| Isochamanetin (10) | N.D | N.D | 19.79 (54.65) 8.46−26.28 | >25.0 (>69.03) | 24.69 (68.18) 17.84−27.52 |
| Dichamanetin (11) | 15.78 (33.70) 14.37−17.33 | 23.59 (50.38) 17.21−32.35 | >25.0 (>53.40) | 18.99 (40.56) 11.98−26.08 | >25.0 (>53.40) |
| Mixture b of uvarinol (12) + isouvarinol (13) | 9.74 7.90−12.01 | >25.0 | >25.0 | 17.31 14.87−20.14 | >25.0 |
| Doxorubicin c | 0.04 (0.07) 0.03–0.05 | 3.08 (5.67) 1.52–6.27 | 2.05 (3.77) 1.34–3.16 | 0.85 (1.56) 0.59−1.24 | 3.19 (5.87) 1.89−5.40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, E.V.; Soares, L.d.N.; Chaar, J.d.S.; Silva, V.R.; Santos, L.d.S.; Koolen, H.H.F.; Silva, F.M.A.d.; Tavares, J.F.; Zengin, G.; Soares, M.B.P.; et al. Benzylated Dihydroflavones and Isoquinoline-Derived Alkaloids from the Bark of Diclinanona calycina (Annonaceae) and Their Cytotoxicities. Molecules 2021, 26, 3714. https://doi.org/10.3390/molecules26123714
Costa EV, Soares LdN, Chaar JdS, Silva VR, Santos LdS, Koolen HHF, Silva FMAd, Tavares JF, Zengin G, Soares MBP, et al. Benzylated Dihydroflavones and Isoquinoline-Derived Alkaloids from the Bark of Diclinanona calycina (Annonaceae) and Their Cytotoxicities. Molecules. 2021; 26(12):3714. https://doi.org/10.3390/molecules26123714
Chicago/Turabian StyleCosta, Emmanoel V., Liviane do N. Soares, Jamal da Silva Chaar, Valdenizia R. Silva, Luciano de S. Santos, Hector H. F. Koolen, Felipe M. A. da Silva, Josean F. Tavares, Gokhan Zengin, Milena B. P. Soares, and et al. 2021. "Benzylated Dihydroflavones and Isoquinoline-Derived Alkaloids from the Bark of Diclinanona calycina (Annonaceae) and Their Cytotoxicities" Molecules 26, no. 12: 3714. https://doi.org/10.3390/molecules26123714
APA StyleCosta, E. V., Soares, L. d. N., Chaar, J. d. S., Silva, V. R., Santos, L. d. S., Koolen, H. H. F., Silva, F. M. A. d., Tavares, J. F., Zengin, G., Soares, M. B. P., & Bezerra, D. P. (2021). Benzylated Dihydroflavones and Isoquinoline-Derived Alkaloids from the Bark of Diclinanona calycina (Annonaceae) and Their Cytotoxicities. Molecules, 26(12), 3714. https://doi.org/10.3390/molecules26123714