
Further Validation of Quantum Crystallography Approaches


Abstract
:1. Introduction
2. Materials and Methods
2.1. Crystallographic Data
2.2. Multipole Model Refinement
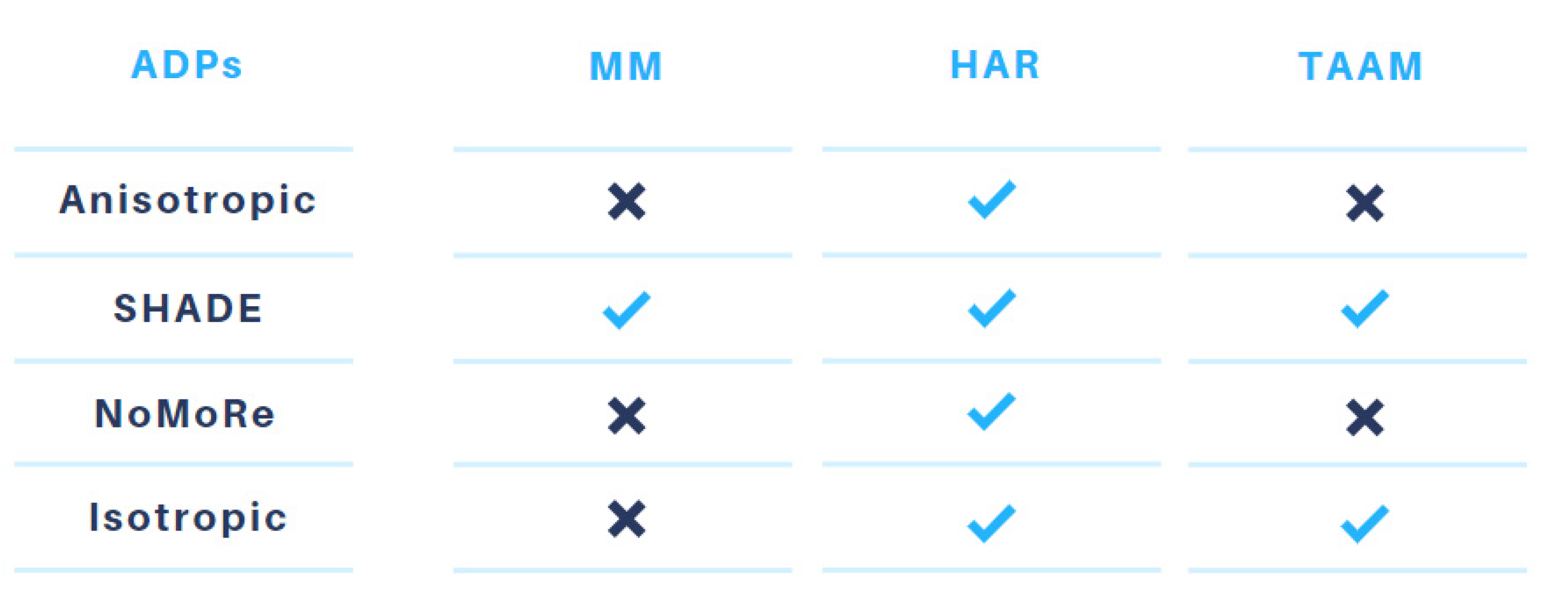
2.3. Hirshfeld Atom Refinement and the Transferable Aspherical Atom Model
2.4. HAR with ADPs from the NoMoRe (Normal Mode Refinement) Method
2.5. ADP Analysis
2.6. Theoretical Computations
3. Results and Discussion
3.1. Refinement Models
3.2. Validation of Refinement Models
3.2.1. Agreement Factors and Residual Density
3.2.2. Geometric Parameters
3.2.3. Lattice Energy
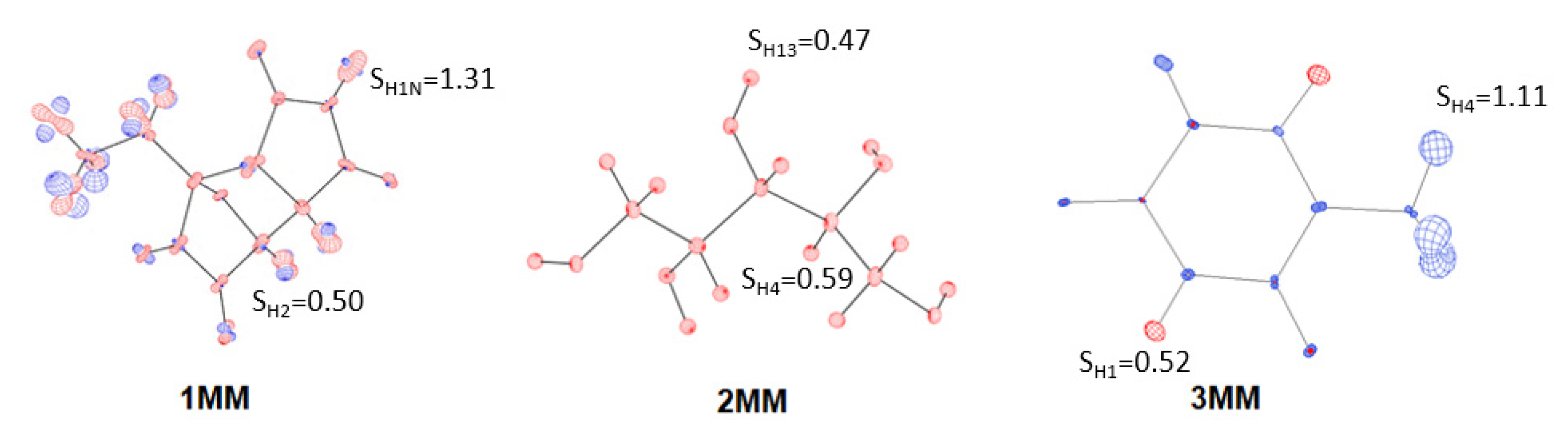
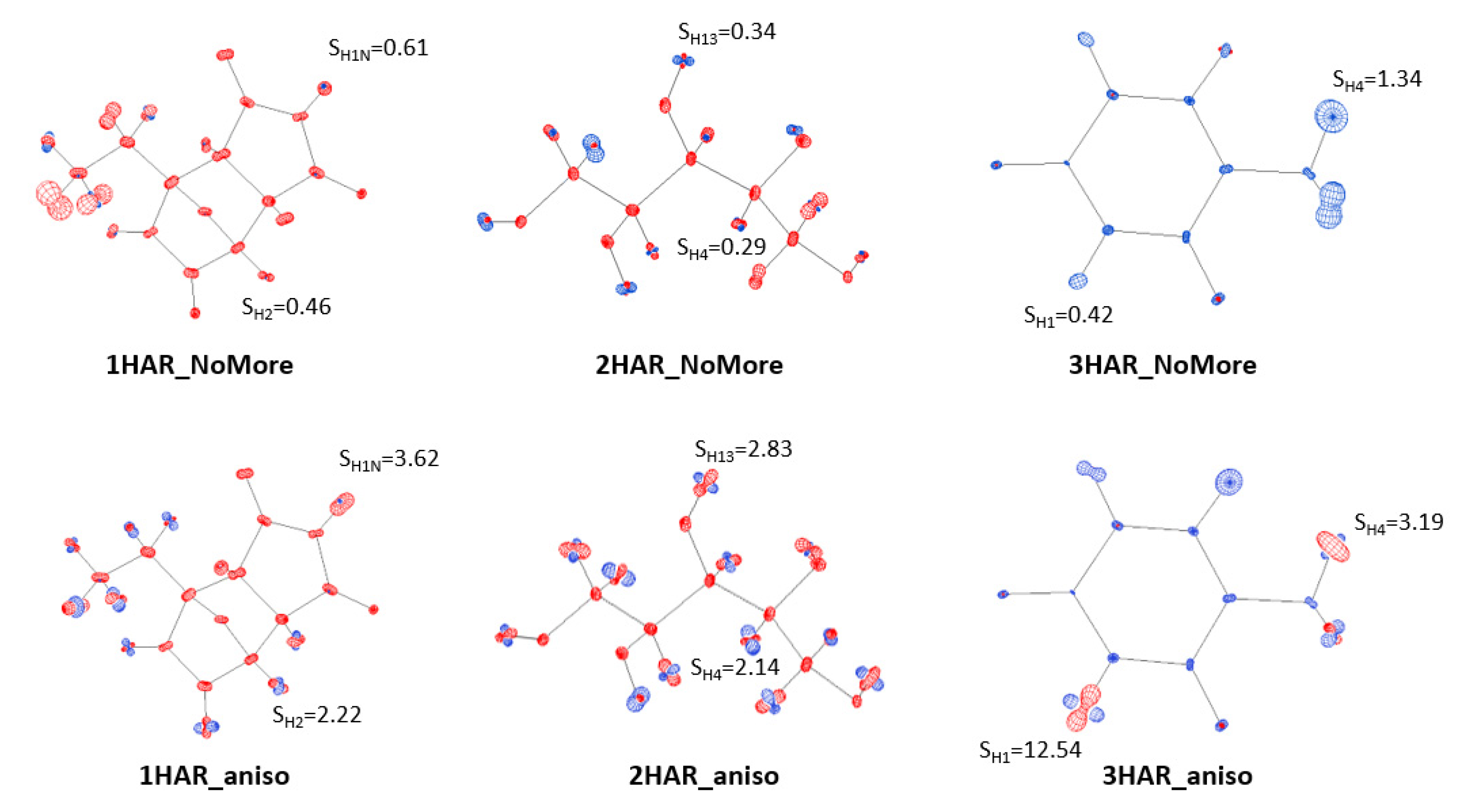
3.3. Results of H Atom ADP Estimation
3.4. Influence of Data Resolution on the Final Results
3.4.1. Geometric Analysis
3.4.2. ADP Analysis
3.4.3. Analysis of Residual Density
4. Conclusions
- According to agreement/discrepancy factors, all methods lead to reasonable models of electron density (see Section 3.2.1).
- Analysis of geometrical parameters revealed that HAR better supplies valence angles closer to the neutron values, whereas bonds, particularly with H atoms, seem to be better described by MM and TAAM (at least in the case of the studied compounds). This may result from restraints applied in those two models. Similar results were also obtained for lattice energies (see Section 3.2.2).
- The HAR model requires restraints to the X–H bonds (see Section 3.2.2).
- Validation of the used models presented in this work revealed that the application of particular treatments of H atoms may have a significant influence on the final results. These effects include:
- The NoMoRe approach may be indicated as the superior method of treatment of hydrogen atom thermal motion (see Section 3.2.2).
- Isotropic refinement of H atoms in HAR led to some of the worst geometric modelling results, whereas isotropic refinement of H atoms in TAAM refinement supplied similar results to the application of SHADE (see Section 3.2.2).
- Anisotropic and NoMoRe approaches mostly resulted in lattice energies closer to the reference neutron values (see Section 3.2.3).
- Finally, we analysed model changes based on refinement against low-resolution Mo Kα and Cu Kα X-ray diffraction data. Results obtained with both wavelengths led to reliable geometry of the final structures (see Section 3.4.1); however, some systematic effects were observed in the ADP values:
- ADPs of heavy atoms obtained with Mo Kα X-ray diffraction data were systematically closer to the ADPs obtained from neutron diffraction, and smaller than those obtained with Cu Kα data (see Section 3.4.2).
- H atoms’ ADP values obtained with Cu Kα data were closer to the neutron ADP values than those obtained with Mo Kα data (0.8 Å) (see Section 3.4.2).
- A better description of ADP values was also reflected in a better fit of the model to the experimental electron density (see Section 3.4.3).
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Genoni, A.; Bučinský, L.; Claiser, N.; Contreras-García, J.; Dittrich, B.; Dominiak, P.M.; Espinosa, E.; Gatti, C.; Giannozzi, P.; Gillet, J.-M.; et al. Quantum Crystallography: Current Developments and Future Perspectives. Chem. A Eur. J. 2018, 24, 10881–10905. [Google Scholar] [CrossRef] [Green Version]
- Hansen, N.K.; Coppens, P. Testing Aspherical Atom Refinements on Small-Molecule Data Sets. Acta Crystallogr. A 1978, 34, 909–921. [Google Scholar] [CrossRef]
- Jarzembska, K.N.; Dominiak, P.M. New Version of the Theoretical Databank of Transferable Aspherical Pseudoatoms, UBDB2011—Towards Nucleic Acid Modelling. Acta Crystallogr. A 2012, 68, 139–147. [Google Scholar] [CrossRef]
- Jelsch, C.; Pichon-Pesme, V.; Lecomte, C.; Aubry, A. Transferability of Multipole Charge-Density Parameters: Application to Very High Resolution Oligopeptide and Protein Structures. Acta Crystallogr. D 1998, 54, 1306–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittrich, B.; Munshi, P.; Spackman, M.A. Invariom-Model Refinement of l-Valinol. Acta Crystallogr. C 2006, 62, o633–o635. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, B.; Hübschle, C.B.; Pröpper, K.; Dietrich, F.; Stolper, T.; Holstein, J. The Generalized Invariom Database (GID). Acta Crystallogr. B 2013, 69, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Bąk, J.M.; Domagała, S.; Hübschle, C.; Jelsch, C.; Dittrich, B.; Dominiak, P.M. Verification of Structural and Electrostatic Properties Obtained by the Use of Different Pseudoatom Databases. Acta Crystallogr. A 2011, 67, 141–153. [Google Scholar] [CrossRef]
- Domagała, S.; Fournier, B.; Liebschner, D.; Guillot, B.; Jelsch, C. An Improved Experimental Databank of Transferable Multipolar Atom Models—ELMAM2. Construction Details and Applications. Acta Crystallogr. A 2012, 68, 337–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capelli, S.C.; Bürgi, H.-B.; Dittrich, B.; Grabowsky, S.; Jayatilaka, D. Hirshfeld Atom Refinement. IUCrJ 2014, 1, 361–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoser, A.A.; Madsen, A.Ø. Dynamic Quantum Crystallography: Lattice-Dynamical Models Refined against Diffraction Data. I. Theory. Acta Crystallogr. A 2016, 72, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Hoser, A.A.; Madsen, A.Ø. Dynamic Quantum Crystallography: Lattice-Dynamical Models Refined against Diffraction Data. II. Applications to l-Alanine, Naphthalene and Xylitol. Acta Crystallogr A 2017, 73, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Wanat, M.; Malinska, M.; Gutmann, M.J.; Cooper, R.I.; Wozniak, K. HAR, TAAM and BODD Refinements of Model Crystal Structures Using Cu Kα and Mo Kα X-ray Diffraction Data. Acta Crystallogr B 2021, 77. [Google Scholar] [CrossRef]
- Köhler, C.; Lübben, J.; Krause, L.; Hoffmann, C.; Herbst-Irmer, R.; Stalke, D. Comparison of Different Strategies for Modelling Hydrogen Atoms in Charge Density Analyses. Acta Crystallogr. B 2019, 75, 434–441. [Google Scholar] [CrossRef] [Green Version]
- Malaspina, L.A.; Hoser, A.A.; Edwards, A.J.; Woińska, M.; Turner, M.J.; Price, J.R.; Sugimoto, K.; Nishibori, E.; Bürgi, H.-B.; Jayatilaka, D.; et al. Hydrogen Atoms in Bridging Positions from Quantum Crystallographic Refinements: Influence of Hydrogen Atom Displacement Parameters on Geometry and Electron Density. CrystEngComm 2020, 22, 4778–4789. [Google Scholar] [CrossRef]
- Orben, C.M.; Dittrich, B. Hydrogen ADPs with Cu Kα Data? Invariom and Hirshfeld Atom Modelling of Fluconazole. Acta Crystallogr. C 2014, 70, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, B.; Weber, M.; Kalinowski, R.; Grabowsky, S.; Hübschle, C.B.; Luger, P. How to Easily Replace the Independent Atom Model—The Example of Bergenin, a Potential Anti-HIV Agent of Traditional Asian Medicine. Acta Crystallogr. B 2009, 65, 749–756. [Google Scholar] [CrossRef]
- Woinska, M.; Wanat, M.; Taciak, P.; Pawinski, T.; Minor, W.; Wozniak, K. Energetics of Interactions in the Solid State of 2-Hydroxy-8-X-Quinoline Derivatives (X = Cl, Br, I, S-Ph): Comparison of Hirshfeld Atom, X-ray Wavefunction and Multipole Refinements. IUCrJ 2019, 6, 868–883. [Google Scholar] [CrossRef] [Green Version]
- Woińska, M.; Jayatilaka, D.; Dittrich, B.; Flaig, R.; Luger, P.; Woźniak, K.; Dominiak, P.M.; Grabowsky, S. Validation of X-ray Wavefunction Refinement. ChemPhysChem 2017, 18, 3334–3351. [Google Scholar] [CrossRef] [PubMed]
- Woińska, M.; Jayatilaka, D.; Spackman, M.A.; Edwards, A.J.; Dominiak, P.M.; Woźniak, K.; Nishibori, E.; Sugimoto, K.; Grabowsky, S. Hirshfeld Atom Refinement for Modelling Strong Hydrogen Bonds. Acta Crystallogr. A 2014, 70, 483–498. [Google Scholar] [CrossRef]
- Chęcińska, L.; Morgenroth, W.; Paulmann, C.; Jayatilaka, D.; Dittrich, B. A Comparison of Electron Density from Hirshfeld-Atom Refinement, X-ray Wavefunction Refinement and Multipole Refinement on Three Urea Derivatives. CrystEngComm 2013, 15, 2084–2090. [Google Scholar] [CrossRef]
- Dittrich, B.; Sze, E.; Holstein, J.J.; Hübschle, C.B.; Jayatilaka, D. Crystal-Field Effects in l-Homoserine: Multipoles versus Quantum Chemistry. Acta Crystallogr. A 2012, 68, 435–442. [Google Scholar] [CrossRef]
- Hickstein, D.D.; Cole, J.M.; Turner, M.J.; Jayatilaka, D. Modeling Electron Density Distributions from X-ray Diffraction to Derive Optical Properties: Constrained Wavefunction versus Multipole Refinement. J. Chem. Phys. 2013, 139, 064108. [Google Scholar] [CrossRef] [PubMed]
- Malinska, M.; Kieliszek, A.; Kozioł, A.E.; Mirosław, B.; Woźniak, K. Interplay between Packing, Dimer Interaction Energy and Morphology in a Series of Tricyclic Imide Crystals. Acta Crystallogr. B 2020, 76, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Madsen, A.Ø.; Sørensen, H.O.; Flensburg, C.; Stewart, R.F.; Larsen, S. Modeling of the Nuclear Parameters for H Atoms in X-ray Charge-Density Studies. Acta Crystallogr. A 2004, 60, 550–561. [Google Scholar] [CrossRef]
- Madsen, A.Ø.; Mason, S.; Larsen, S. A Neutron Diffraction Study of Xylitol: Derivation of Mean Square Internal Vibrations for H Atoms from a Rigid-Body Description. Acta Crystallogr. B 2003, 59, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMullan, R.K.; Craven, B.M. Crystal Structure of 1-Methyluracil from Neutron Diffraction at 15, 60 and 123 K. Acta Crystallogr. B 1989, 45, 270–276. [Google Scholar] [CrossRef]
- Jelsch, C.; Guillot, B.; Lagoutte, A.; Lecomte, C. Advances in Protein and Small-Molecule Charge-Density Refinement Methods Using MoPro. J. Appl. Crystallogr. 2005, 38, 38–54. [Google Scholar] [CrossRef] [Green Version]
- Madsen, A.Ø. SHADE Web Server for Estimation of Hydrogen Anisotropic Displacement Parameters. J. Appl. Crystallogr. 2006, 39, 757–758. [Google Scholar] [CrossRef] [Green Version]
- Madsen, A.Ø.; Hoser, A.A. SHADE3 Server: A Streamlined Approach to Estimate H-Atom Anisotropic Displacement Parameters Using Periodic Ab Initio Calculations or Experimental Information. J. Appl. Crystallogr. 2014, 47, 2100–2104. [Google Scholar] [CrossRef]
- Allen, F.H.; Bruno, I.J. Bond Lengths in Organic and Metal-Organic Compounds Revisited: X—H Bond Lengths from Neutron Diffraction Data. Acta Crystallogr. B 2010, 66, 380–386. [Google Scholar] [CrossRef]
- Volkov, A.; Macchi, P.; Farrugia, L.; Gatti, C.; Mallinson, P.R.; Richter, T.; Koritsanszky, T. XD2016—A Computer Program Package for Multipole Refinement, Topological Analysis of Charge Densities and Evaluation of Intermolecular Energies from Experimental and Theoretical Structure Factors. 2016. Available online: https://www.chem.gla.ac.uk/~louis/xd-home/avail.html (accessed on 14 June 2021).
- Volkov, A.; Li, X.; Koritsanszky, T.; Coppens, P. Ab Initio Quality Electrostatic Atomic and Molecular Properties Including Intermolecular Energies from a Transferable Theoretical Pseudoatom Databank. J. Phys. Chem. A 2004, 108, 4283–4300. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Woińska, M.; Grabowsky, S.; Dominiak, P.M.; Woźniak, K.; Jayatilaka, D. Hydrogen Atoms Can Be Located Accurately and Precisely by X-ray Crystallography. Sci. Adv. 2016, 2, e1600192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J. CRYSTAL17. In CRYSTAL17 User’s Manual Torino: University of Torino; 2017; Available online: https://www.crystal.unito.it/how-to-cite.php (accessed on 14 June 2021).
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S. Quantum-Mechanical Condensed Matter Simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Bučinský, L.; Jayatilaka, D.; Grabowsky, S. Relativistic Quantum Crystallography of Diphenyl- and Dicyanomercury. Theoretical Structure Factors and Hirshfeld Atom Refinement. Acta Crystallogr. A 2019, 75, 705–717. [Google Scholar] [CrossRef]
- Whitten, A.E.; Spackman, M.A. Anisotropic Displacement Parameters for H Atoms Using an ONIOM Approach. Acta Crystallogr. B 2006, 62, 875–888. [Google Scholar] [CrossRef] [Green Version]
- Mroz, D.; George, J.; Kremer, M.; Wang, R.; Englert, U.; Dronskowski, R. A New Tool for Validating Theoretically Derived Anisotropic Displacement Parameters with Experiment: Directionality of Prolate Displacement Ellipsoids. CrystEngComm 2019, 21, 6396–6404. [Google Scholar] [CrossRef]
- Hummel, W.; Hauser, J.; Bürgi, H.-B. PEANUT: Computer Graphics Program to Represent Atomic Displacement Parameters. J. Mol. Graph. 1990, 8, 214–220. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Civalleri, B.; Zicovich-Wilson, C.M.; Valenzano, L.; Ugliengo, P. B3LYP Augmented with an Empirical Dispersion Term (B3LYP-D*) as Applied to Molecular Crystals. CrystEngComm 2008, 10, 405–410. [Google Scholar] [CrossRef]
- Henn, J.; Meindl, K. Two Common Sources of Systematic Errors in Charge Density Studies. Int. J. Mater. Chem. Phys. 2015, 1, 417–430. [Google Scholar]
- Sanjuan-Szklarz, W.F.; Woińska, M.; Domagala, S.; Dominiak, P.M.; Grabowsky, S.; Jayatilaka, D.; Gutmann, M.; Woźniak, K. On the Accuracy and Precision of X-ray and Neutron Diffraction Results as a Function of Resolution and the Electron Density Model. IUCrJ 2020, 7, 920–933. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Parameters | MM | HAR Aniso | HAR SHADE | HAR NoMoRe | HAR Iso | TAAM SHADE | TAAM Iso |
|---|---|---|---|---|---|---|---|---|
| 1 | R(F > 2σ(F)) | 0.013 | 0.015 | 0.015 | 0.015 | 0.017 | 0.015 | 0.018 |
| wR(F2) | 0.043 | 0.045 | 0.045 | 0.045 | 0.051 | 0.050 | 0.057 | |
| # of reflections | 9849 | 9828 | 9826 | 9828 | 9828 | 9849 | 9849 | |
| # of fit parameters | 160 | 226 | 160 | 160 | 160 | 160 | 171 | |
| chi2 | n/a | 2.35 | 2.39 | 2.41 | 2.94 | n/a | n/a | |
| Goodness of fit | 1.35 | 1.53 | 1.55 | 1.55 | 1.72 | 1.58 | 1.77 | |
| Δρmax | 0.22 | 0.20 | 0.22 | 0.21 | 0.22 | 0.33 | 0.33 | |
| Δρmin | −0.16 | −0.15 | −0.15 | −0.15 | −0.16 | −0.21 | −0.33 | |
| MM | HAR aniso | HAR SHADE | HAR NoMoRe | HAR iso | TAAM SHADE | TAAM iso | ||
| 2 | R(F > 2σ(F)) | 0.021 | 0.021 | 0.021 | 0.021 | 0.022 | 0.018 | 0.023 |
| wR(F2) | 0.029 | 0.030 | 0.031 | 0.031 | 0.034 | 0.035 | 0.038 | |
| # of reflections | 9779 | 9776 | 9776 | 9776 | 9776 | 9779 | 9779 | |
| # of fit parameters | 127 | 199 | 127 | 127 | 127 | 127 | 139 | |
| chi2 | n/a | 0.50 | 0.52 | 0.51 | 0.62 | n/a | n/a | |
| Goodness of fit | 0.67 | 0.71 | 0.72 | 0.72 | 0.79 | 0.82 | 0.89 | |
| Δρmax | 0.24 | 0.16 | 0.15 | 0.15 | 0.15 | 0.24 | 0.32 | |
| Δρmin | −0.27 | −0.19 | −0.19 | −0.19 | −0.19 | −0.32 | −0.46 | |
| MM | HAR aniso | HAR SHADE | HAR NoMoRe | HAR iso | TAAM SHADE | TAAM iso | ||
| 3 | R (F > 2σ(F)) | 0.037 | 0.038 | 0.039 | 0.039 | 0.039 | 0.051 | 0.031 |
| wR(F2) | 0.057 | 0.060 | 0.061 | 0.061 | 0.066 | 0.074 | 0.070 | |
| # of reflections | 3113 | 3113 | 3113 | 3113 | 3113 | 3113 | 3113 | |
| # of fit parameters | 56 | 88 | 66 | 66 | 66 | 55 | 60 | |
| chi2 | n/a | 1.86 | 1.91 | 1.93 | 1.91 | n/a | n/a | |
| Goodness of fit | 1.24 | 1.36 | 1.38 | 1.39 | 1.38 | 1.56 | 1.51 | |
| Δρmax | 0.42 | 0.16 | 0.17 | 0.17 | 0.17 | 0.49 | 0.44 | |
| Δρmin | −0.33 | −0.17 | −0.17 | −0.17 | −0.17 | −0.42 | −0.29 |
| Model | 1 | 2 | 3 |
|---|---|---|---|
| MM | 1.28 | 11.63 | 1.72 |
| IAM | 3.05 | 12.35 | 0.25 |
| HAR_aniso | 2.75 | 13.65 | 1.70 |
| HAR_Shade | 2.74 | 14.16 | 2.37 |
| HAR_NoMoRe | 2.46 | 14.44 | 2.08 |
| HAR_iso | 2.54 | 15.34 | 2.38 |
| TAAM_Shade | 1.39 | 11.67 | 2.94 |
| TAAM_iso | 1.33 | 11.37 | 2.94 |
| Compound | MM | HAR_Aniso | HAR_SHADE | HAR_NoMoRe | TAAM_SHADE |
|---|---|---|---|---|---|
| 1 | 0.7 (2) | 1.4 (3) | 0.7 (1) | 0.6 (1) | 0.7 (1) |
| 2 | 0.8 (2) | 2.2 (4) | 0.8 (2) | 0.4 (1) | 0.8 (2) |
| 3 | 0.9 (4) | 4 (2) | 0.5 (1) | 0.6 (2) | 0.7 (3) |
| Compound | HAR_Aniso | HAR_SHADE | TAAM_SHADE |
|---|---|---|---|
| 1 | 1.43 | 0.70 | 0.72 |
| 1-cutoff | 2.55 | 0.79 | 0.87 |
| 1-Cu Kα | 2.13 | 0.96 | 1.14 |
| 2 | 2.16 | 0.78 | 0.79 |
| 2-cutoff | 11.18 * | 4.2 | 4.21 |
| 2-Cu Kα | 10.19 * | 1.19 | 1.34 |
| 3 | 4.14 | 0.47 | 0.73 |
| 3-cutoff | 6.64 | 2.22 | 3.12 |
| 3-Cu Kα | 5.80 | 0.74 | 1.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wanat, M.; Malinska, M.; Hoser, A.A.; Woźniak, K. Further Validation of Quantum Crystallography Approaches. Molecules 2021, 26, 3730. https://doi.org/10.3390/molecules26123730
Wanat M, Malinska M, Hoser AA, Woźniak K. Further Validation of Quantum Crystallography Approaches. Molecules. 2021; 26(12):3730. https://doi.org/10.3390/molecules26123730
Chicago/Turabian StyleWanat, Monika, Maura Malinska, Anna A. Hoser, and Krzysztof Woźniak. 2021. "Further Validation of Quantum Crystallography Approaches" Molecules 26, no. 12: 3730. https://doi.org/10.3390/molecules26123730
APA StyleWanat, M., Malinska, M., Hoser, A. A., & Woźniak, K. (2021). Further Validation of Quantum Crystallography Approaches. Molecules, 26(12), 3730. https://doi.org/10.3390/molecules26123730