
Enantioenriched Positive Allosteric Modulators Display Distinct Pharmacology at the Dopamine D1 Receptor



Abstract
:1. Introduction
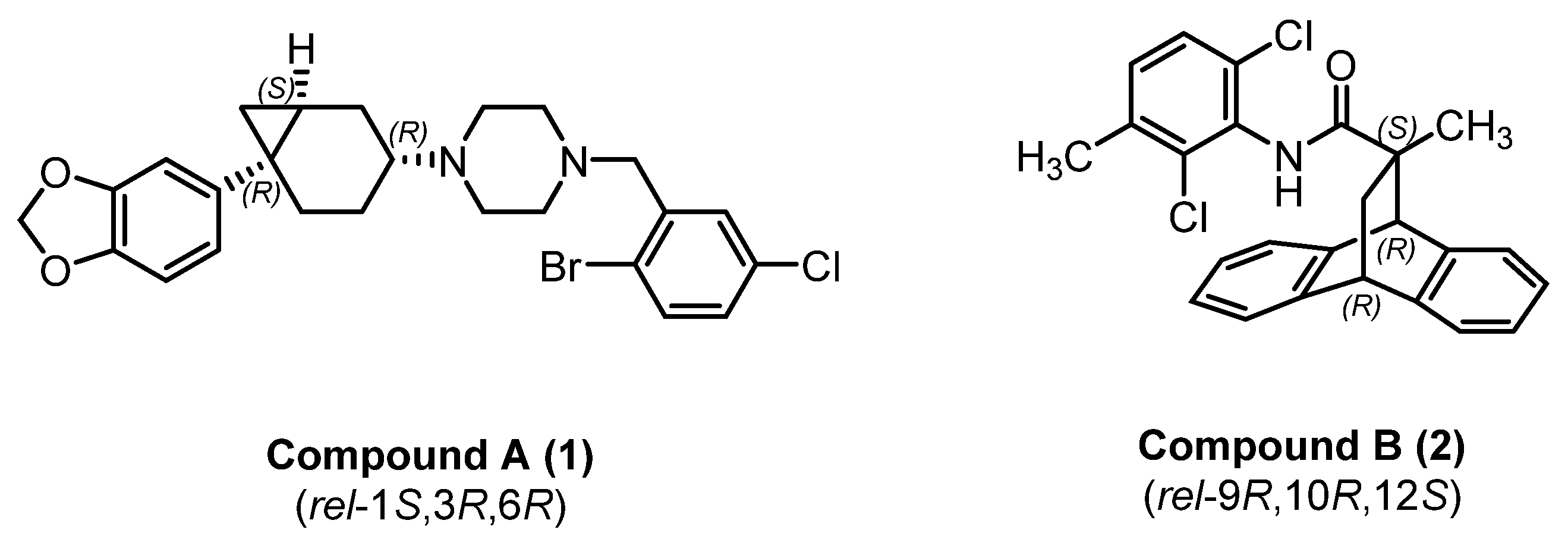
2. Results and Discussions
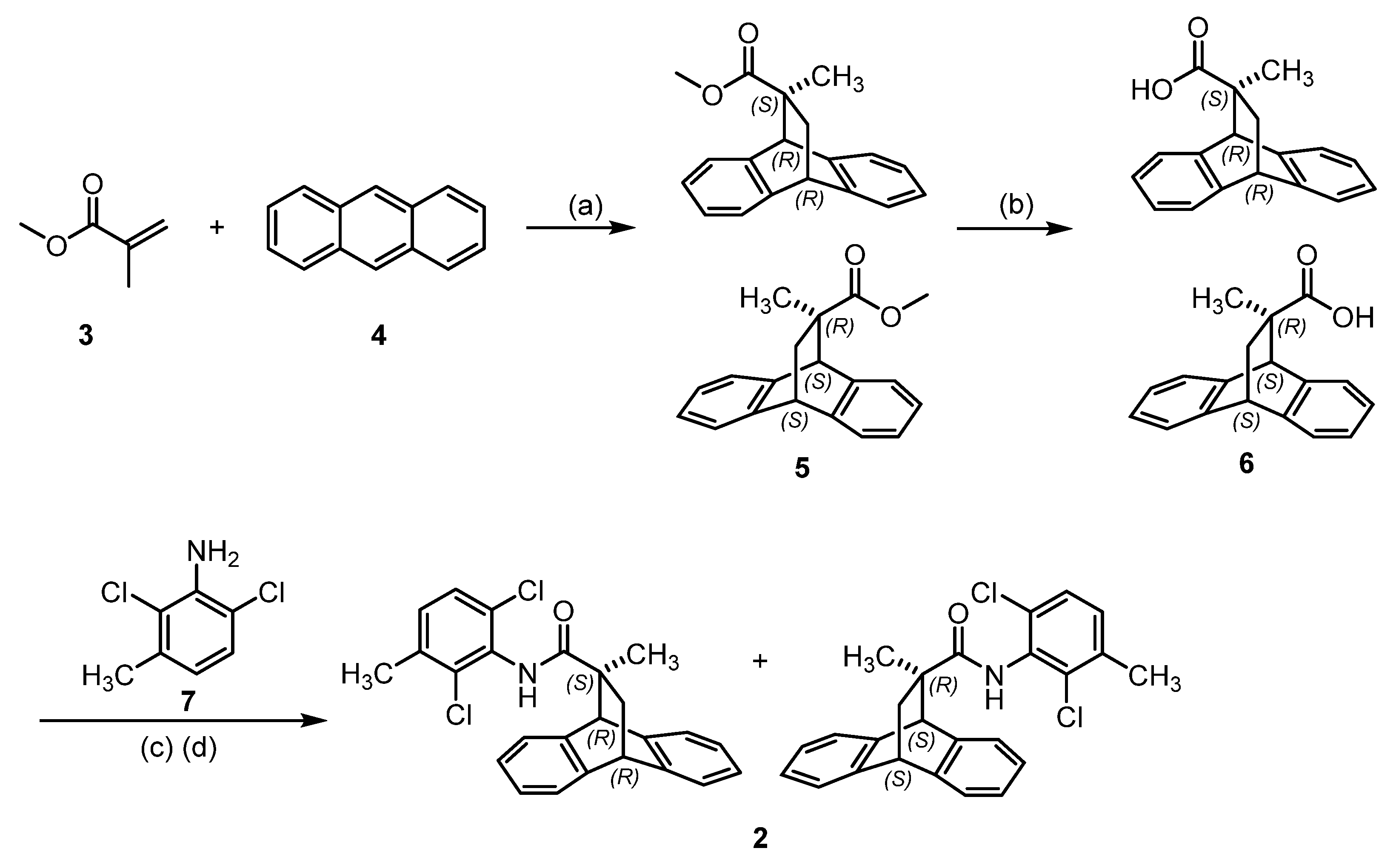
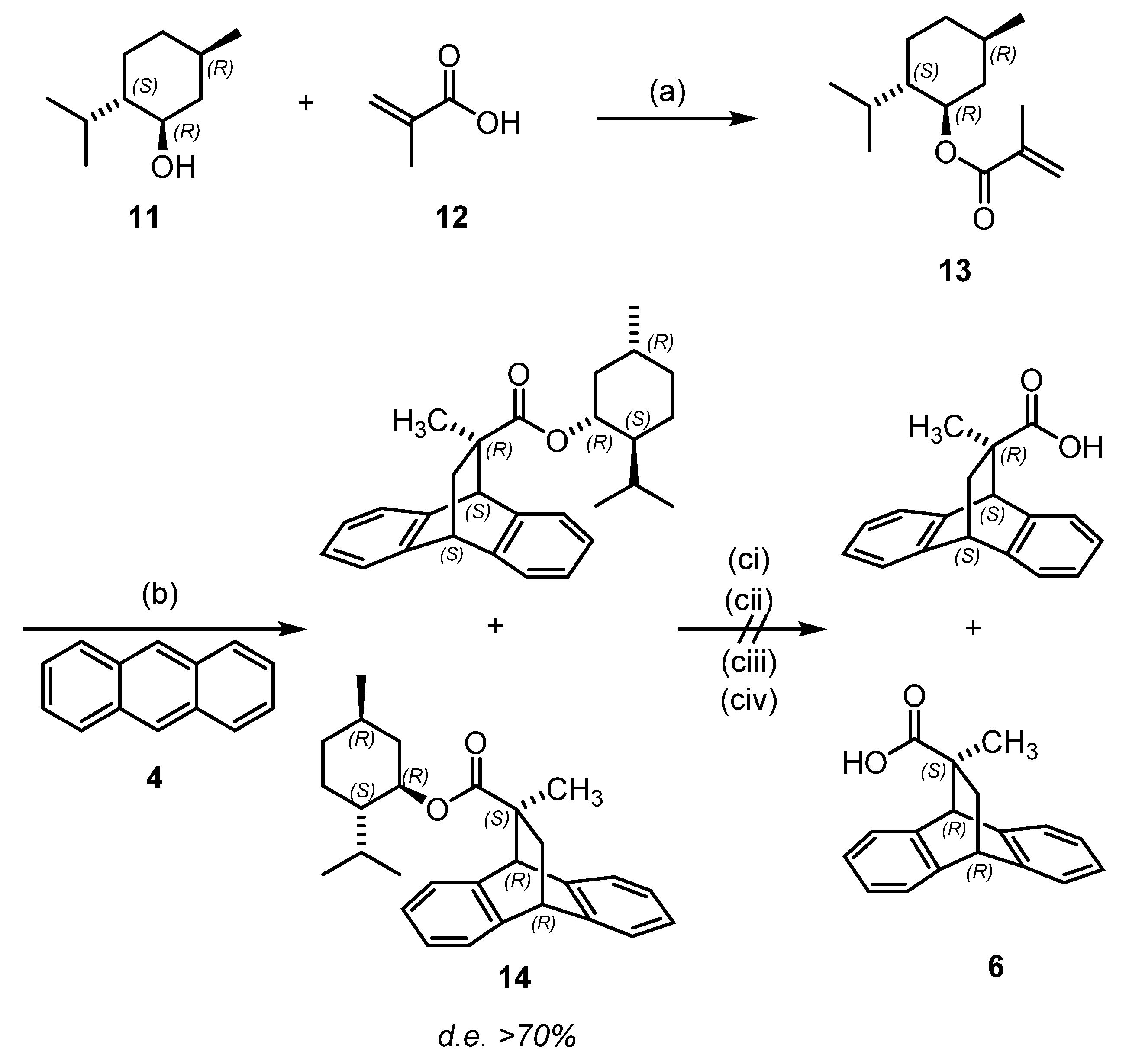
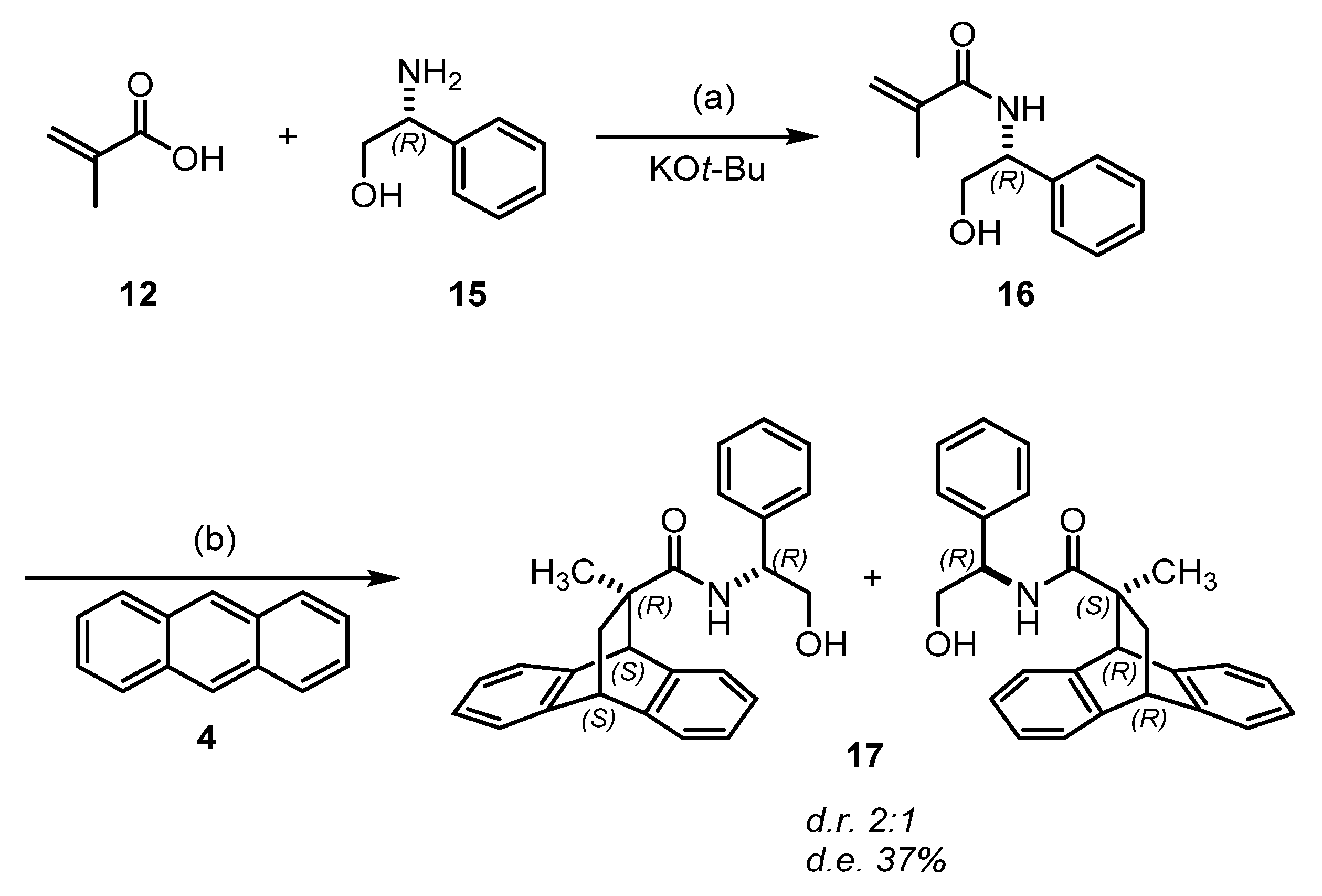
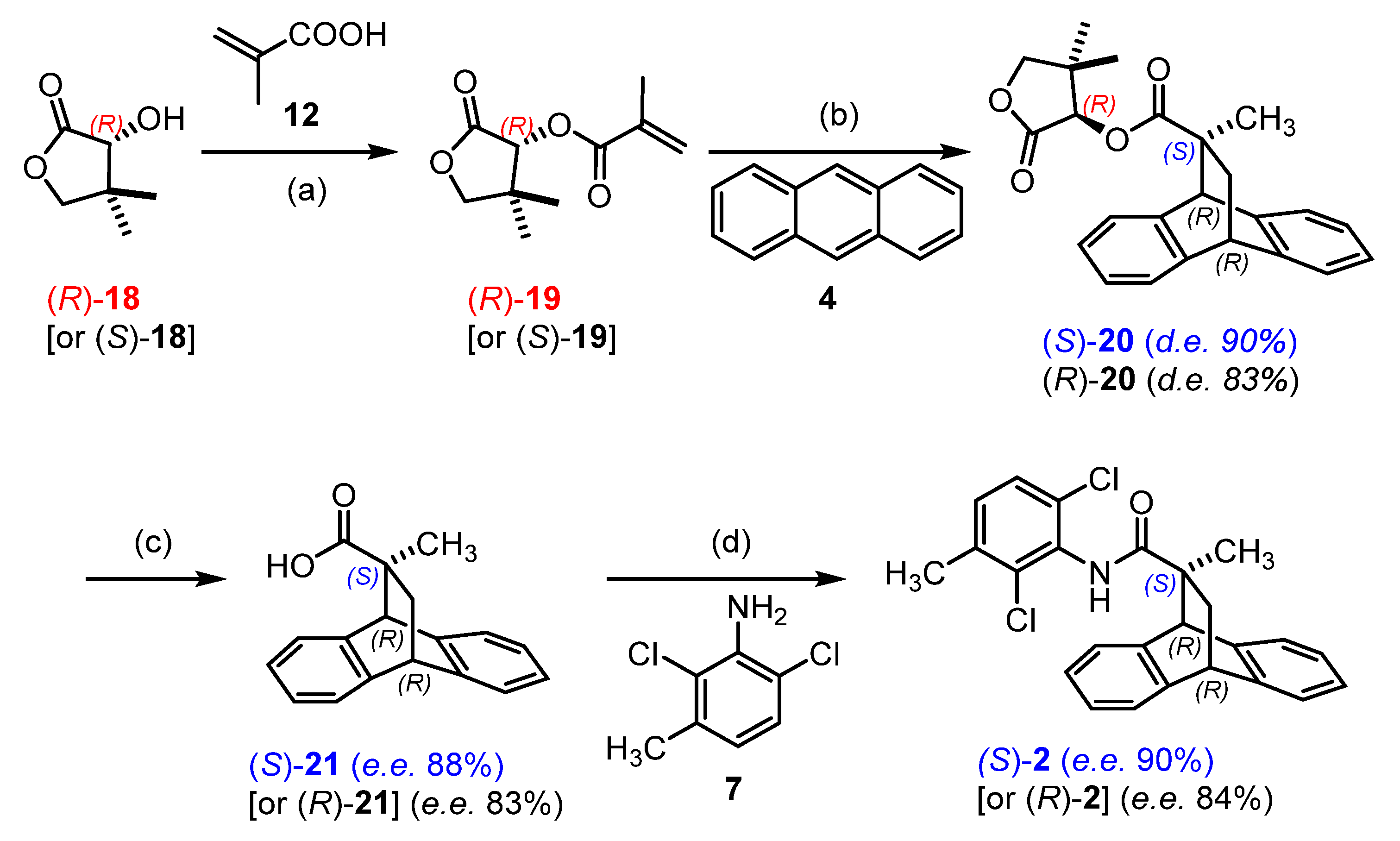
2.1. Chemistry
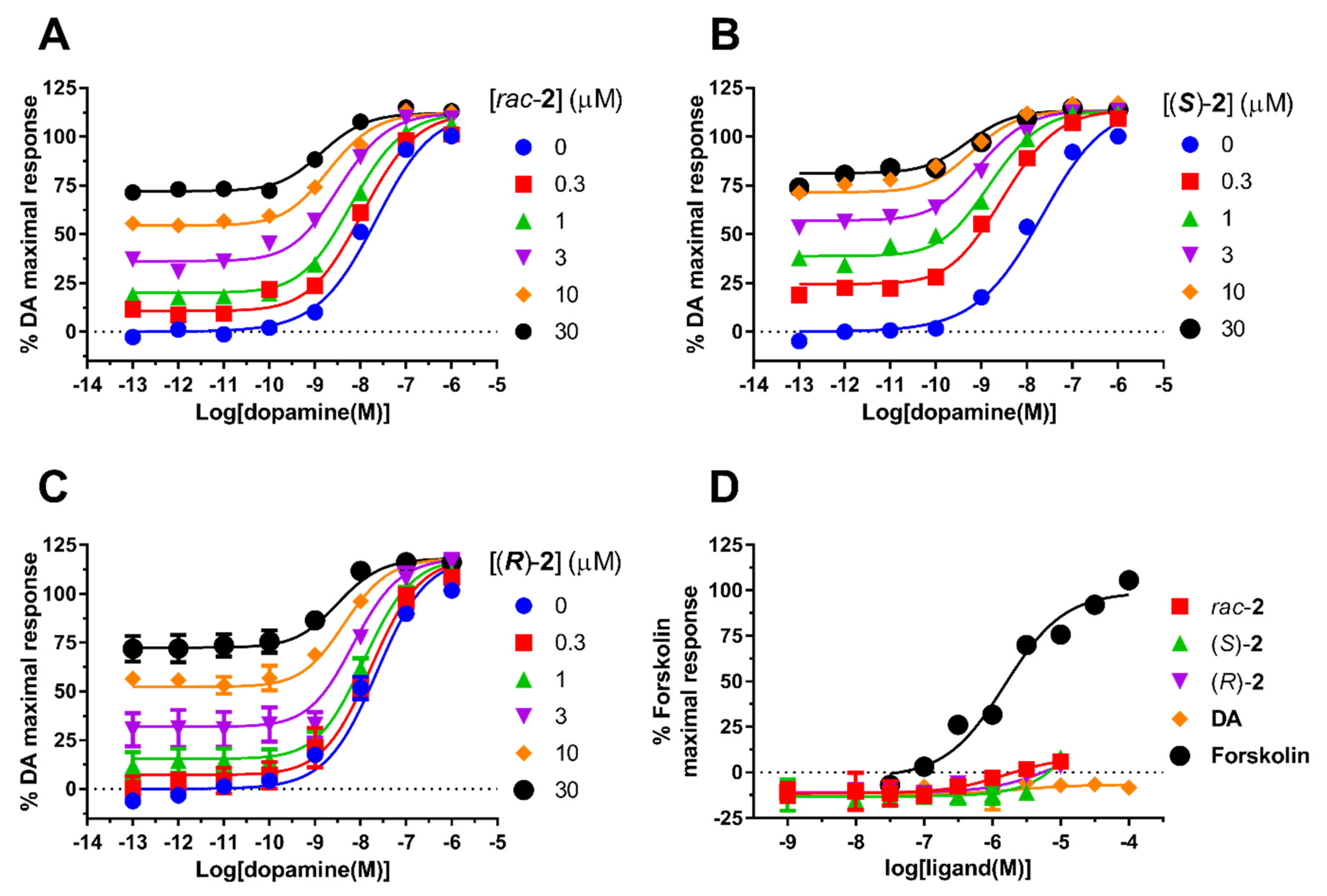
2.2. Pharmacology
3. Experimental
3.1. General
3.2. Chemistry
3.2.1. General Procedure A for Steglich Esterification
3.2.2. General Procedure B for TiCl4-Catalysed Diels-Alder Cycloaddition
3.2.3. General Procedure C for Alkaline Ester Hydrolysis
3.2.4. General Procedure D for Acyl Halide Formation and Nucleophilic Substitution















3.3. Pharmacological Characterisation
3.3.1. Materials
3.3.2. General Cell Culture
Cell Culture and Transfection for cAMP Assay
3.3.3. cAMP Assay Measurement
3.3.4. Data Analysis of Allsotery
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ibrahim, H.M.; Tamminga, C.A. Schizophrenia: Treatment targets beyond monoamine systems. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 189–209. [Google Scholar] [CrossRef]
- Van Os, J.; Kapur, S. Schizophrenia. Lancet 2009, 374, 635–645. [Google Scholar] [CrossRef]
- Rosen, W.G.; Mohs, R.C.; Johns, C.A.; Small, N.S.; Kendler, K.S.; Horvath, T.B.; Davis, K.L. Positive and negative symptoms in schizophrenia. Psychiatry Res. 1984, 13, 277–284. [Google Scholar] [CrossRef]
- Bowie, C.R.; Harvey, P.D. Cognitive deficits and functional outcome in schizophrenia. Neuropsychiatr. Dis. Treat. 2006, 2, 531–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Tuathaigh, C.M.P.; Moran, P.M.; Zhen, X.C.; Waddington, J.L. Translating advances in the molecular basis of schizophrenia into novel cognitive treatment strategies. Br. J. Pharmacol. 2017, 174, 3173–3190. [Google Scholar] [CrossRef] [Green Version]
- Leucht, S.; Corves, C.; Arbter, D.; Engel, R.R.; Li, C.; Davis, J.M. Second-generation versus first-generation antipsychotic drugs for schizophrenia: A meta-analysis. Lancet 2009, 373, 31–41. [Google Scholar] [CrossRef]
- Koster, L.S.; Carbon, M.; Correll, C.U. Emerging drugs for schizophrenia: An update. Expert Opin. Emerg. Drugs 2014, 19, 511–531. [Google Scholar] [CrossRef]
- Curtis, C.E.; D’Esposito, M. Persistent activity in the prefrontal cortex during working memory. Trends Cogn. Sci. 2003, 7, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Davis, K.L.; Kahn, R.S.; Ko, G.; Davidson, M. Dopamine in schizophrenia: A review and reconceptualization. Am. J. Psychiatry 1991, 148, 1474–1486. [Google Scholar] [CrossRef]
- Weinberger, D.R. Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry 1987, 44, 660–669. [Google Scholar] [CrossRef]
- Brozoski, T.J.; Brown, R.M.; Rosvold, H.E.; Goldman, P.S. Cognitive deficit caused by regional depletion of dopamine in prefrontal cortex of rhesus monkey. Science 1979, 205, 929–932. [Google Scholar] [CrossRef] [PubMed]
- Sawaguchi, T.; Goldman-Rakic, P.S. The role of D1-dopamine receptor in working memory: Local injections of dopamine antagonists into the prefrontal cortex of rhesus monkeys performing an oculomotor delayed-response task. J. Neurophysiol. 1994, 71, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Sawaguchi, T.; Goldman-Rakic, P.S. D1 dopamine receptors in prefrontal cortex: Involvement in working memory. Science 1991, 251, 947–950. [Google Scholar] [CrossRef]
- Arnsten, A.F.; Cai, J.X.; Murphy, B.L.; Goldman-Rakic, P.S. Dopamine D1 receptor mechanisms in the cognitive performance of young adult and aged monkeys. Psychopharmacology 1994, 116, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Locke, T.M.; Soden, M.E.; Miller, S.M.; Hunker, A.; Knakal, C.; Licholai, J.A.; Dhillon, K.S.; Keene, C.D.; Zweifel, L.S.; Carlson, E.S. Dopamine D1 receptor–Positive neurons in the lateral nucleus of the cerebellum contribute to cognitive behavior. Biol. Psychiatry 2018. [Google Scholar] [CrossRef] [Green Version]
- Abi-Dargham, A.; Moore, H. Prefrontal DA transmission at D1 receptors and the pathology of schizophrenia. Neuroscientist 2003, 9, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Vijayraghavan, S.; Wang, M.; Birnbaum, S.G.; Williams, G.V.; Arnsten, A.F. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat. Neurosci. 2007, 10, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Mailman, R.; Huang, X.; Nichols, D.E. Parkinson’s disease and D1 dopamine receptors. Curr. Opin. Investig. Drugs 2001, 2, 1582–1591. [Google Scholar]
- Starr, M.S.; Starr, B.S. Seizure promotion by D1 agonists does not correlate with other dopaminergic properties. J. Neural Transm. Parkinson’s Dis. Dement. Sect. 1993, 6, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F.; Girgis, R.R.; Gray, D.L.; Mailman, R.B. Novel Dopamine Therapeutics for Cognitive Deficits in Schizophrenia. Biol. Psychiatry 2017, 81, 67–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amenta, F.; Ferrante, F.; Ricci, A. Pharmacological Characterisation and Autoradiographic Localisation of Dopamine Receptor Subtypes in the Cardiovascular System and in the Kidney. Hypertens. Res. 1995, 18, S23–S27. [Google Scholar] [CrossRef] [Green Version]
- Ryman-Rasmussen, J.P.; Griffith, A.; Oloff, S.; Vaidehi, N.; Brown, J.T.; Goddard, W.A., 3rd; Mailman, R.B. Functional selectivity of dopamine D1 receptor agonists in regulating the fate of internalized receptors. Neuropharmacology 2007, 52, 562–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulwadi, A.G.; Korpinen, C.D.; Mailman, R.B.; Nichols, D.E.; Sit, S.Y.; Taber, M.T. Dinapsoline: Characterization of a D1 dopamine receptor agonist in a rat model of Parkinson’s disease. J. Pharmacol. Exp. Ther. 2001, 296, 338–344. [Google Scholar]
- Bruns, R.F.; Mitchell, S.N.; Wafford, K.A.; Harper, A.J.; Shanks, E.A.; Carter, G.; O’Neill, M.J.; Murray, T.K.; Eastwood, B.J.; Schaus, J.M.; et al. Preclinical profile of a dopamine D1 potentiator suggests therapeutic utility in neurological and psychiatric disorders. Neuropharmacology 2018, 128, 351–365. [Google Scholar] [CrossRef]
- Svensson, K.A.; Heinz, B.A.; Schaus, J.M.; Beck, J.P.; Hao, J.; Krushinski, J.H.; Reinhard, M.R.; Cohen, M.P.; Hellman, S.L.; Getman, B.G.; et al. An allosteric potentiator of the dopamine D1 receptor increases locomotor activity in human D1 knock-in mice without causing stereotypy or tachyphylaxis. J. Pharmacol. Exp. Ther. 2017, 360, 117–128. [Google Scholar] [CrossRef]
- Luderman, K.D.; Conroy, J.L.; Free, R.B.; Southall, N.; Ferrer, M.; Sanchez-Soto, M.; Moritz, A.E.; Willette, B.K.A.; Fyfe, T.J.; Jain, P.; et al. Identification of positive allosteric modulators of the D1 dopamine receptor that act at diverse binding sites. Mol. Pharmacol. 2018, 94, 1197–1209. [Google Scholar] [CrossRef]
- Hall, A.; Provins, L.; Valade, A. Novel strategies to activate the dopamine D1 receptor: Recent advances in orthosteric agonism and positive allosteric modulation. J. Med. Chem. 2019, 62, 128–140. [Google Scholar] [CrossRef]
- Svensson, K.A.; Hao, J.; Bruns, R.F. Positive allosteric modulators of the dopamine D1 receptor: A new mechanism for the treatment of neuropsychiatric disorders. Adv. Pharmacol. 2019, 86, 273–305. [Google Scholar] [CrossRef]
- Luderman, K.D.; Jain, P.; Free, R.B.; Conroy, J.L.; Aubé, J.; Sibley, D.R.; Frankowski, K.J. Development of pyrimidone D1 dopamine receptor positive allosteric modulators. Bioorg. Med. Chem. Lett. 2021, 31, 127696. [Google Scholar] [CrossRef]
- Sun, B.; Feng, D.; Chu, M.L.H.; Fish, I.; Lovera, S.; Sands, Z.A.; Kelm, S.; Valade, A.; Wood, M.; Ceska, T.; et al. Crystal structure of dopamine D1 receptor in complex with G protein and a non-catechol agonist. Nat. Commun. 2021, 12, 3305. [Google Scholar] [CrossRef]
- Sibley, D.R.; Luderman, K.D.; Free, R.B.; Shi, L. Novel Cryo-EM structures of the D1 dopamine receptor unlock its therapeutic potential. Signal Transduct. Target. Ther. 2021, 6, 205. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Xu, P.; Mao, C.; Wang, L.; Krumm, B.; Zhou, X.E.; Huang, S.; Liu, H.; Cheng, X.; Huang, X.P.; et al. Structural insights into the human D1 and D2 dopamine receptor signaling complexes. Cell 2021, 184, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Wilbraham, D.; Biglan, K.M.; Svensson, K.A.; Tsai, M.; Kielbasa, W. Safety, tolerability, and pharmacokinetics of Mevidalen (LY3154207), a centrally acting dopamine D1 receptor-positive allosteric modulator (D1PAM), in healthy subjects. Clin. Pharmacol. Drug Dev. 2021, 10, 393–403. [Google Scholar] [CrossRef]
- Christopoulos, A.; Kenakin, T. G protein-coupled receptor allosterism and complexing. Pharmacol. Rev. 2002, 54, 323–374. [Google Scholar] [CrossRef]
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.A.; Hunihan, L.; Watson, J.; Gentles, R.G.; Hu, S.; Huang, Y.; Bronson, J.; Macor, J.E.; Beno, B.R.; Ferrante, M.; et al. Discovery of D1 dopamine receptor positive allosteric modulators: Characterization of pharmacology and identification of residues that regulate species selectivity. J. Pharmacol. Exp. Ther. 2015, 354, 340–349. [Google Scholar] [CrossRef] [Green Version]
- Leach, K.; Sexton, P.M.; Christopoulos, A. Allosteric GPCR modulators: Taking advantage of permissive receptor pharmacology. Trends Pharmacol. Sci. 2007, 28, 382–389. [Google Scholar] [CrossRef]
- Cabellero, A.G.; Croft, A.K.; Nalli, S.M. Remote aromatic stabilization in radical reactions. Tetrahedron Lett. 2008, 49, 3613–3615. [Google Scholar] [CrossRef] [Green Version]
- Camps, P.; Font-Bardia, M.; Giménez, S.; Pérez, F.; Solans, X.; Soldevilla, N. (R)- and (S)-3-Hydroxy-4,4-dimethyl-1-phenyl-2-pyrrolidinone as chiral auxiliaries in Diels-Alder reactions. Tetrahedron Asymmetry 1999, 10, 3123–3138. [Google Scholar] [CrossRef]
- Brocksom, T.J.; Nakamura, J.; Ferreira, M.L.; Brocksom, U. The Diels-Alder reaction: An update. J. Braz. Chem. Soc. 2001, 12, 597–622. [Google Scholar] [CrossRef]
- Kagan, H.B.; Riant, O. Catalytic asymmetric Diels Alder reactions. Chem. Rev. 1992, 92, 1007–1019. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Snyder, S.A.; Montagnon, T.; Vassilikogiannakis, G. The Diels-Alder reaction in total synthesis. Angew. Chem. 2002, 41, 1668–1698. [Google Scholar] [CrossRef]
- Corey, E.J.; Ensley, H.E. Preparation of an optically active prostaglandin intermediate via asymmetric induction. J. Am. Chem. Soc. 1975, 97, 6908–6909. [Google Scholar] [CrossRef] [PubMed]
- Neises, B.; Steglich, W. Simple method for the esterification of carboxylic acids. Angew. Chem. Int. Ed. Engl. 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Myers, A.G.; Yang, B.H.; Chen, H.; McKinstry, L.; Kopecky, D.J.; Gleason, J.L. Pseudoephedrine as a practical chiral auxiliary for the synthesis of highly enantiomerically enriched carboxylic acids, alcohols, aldehydes, and ketones. J. Am. Chem. Soc. 1997, 119, 6496–6511. [Google Scholar] [CrossRef]
- Kim, B.R.; Lee, H.-G.; Kang, S.-B.; Sung, G.H.; Kim, J.-J.; Park, J.K.; Lee, S.-G.; Yoon, Y.-J. tert-Butoxide-assisted amidation of esters under green conditions. Synthesis 2012, 44, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Poll, T.; Sobczak, A.; Hartmann, H.; Helmchen, G. Diastereoface-discriminative metal coordination in asymmetric synthesis: d-pantolactone as practical chiral auxiliary for Lewis acid catalyzed Diels-Alder reactions. Tetrahedron Lett. 1985, 26, 3095–3098. [Google Scholar] [CrossRef]
- Miyaji, K.; Ohara, Y.; Takahashi, Y.; Tsuruda, T.; Arai, K. Synthesis of Corey lactone via highly stereoselective asymmetric Diels-Alder reaction. Tetrahedron Lett. 1991, 32, 4557–4560. [Google Scholar] [CrossRef]
- Helmchen, G.; Ihrig, K.; Schindler, H. EPC-syntheses via asymmetric Diels-Alder reactions/retro Diels-Alder reactions I: (R)- and (S)-matsutake alcohol. (R)- and (S)-sarcomycin methyl ester. Tetrahedron Lett. 1987, 28, 183–186. [Google Scholar] [CrossRef]
- Pelayo, C.; Diego, M.-T. Synthesis and applications of (R)- and (S)-pantolactone as chiral auxiliaries. Curr. Org. Chem. 2004, 8, 1339–1380. [Google Scholar] [CrossRef]
- Manickam, G.; Sundararajan, G. Asymmetric Diels-Alder and ene reactions promoted by a Ti(IV) complex bearing a C2-symmetric tridentate ligand. Tetrahedron Asymmetry 1999, 10, 2913–2925. [Google Scholar] [CrossRef]
- Jiang, L.I.; Collins, J.; Davis, R.; Lin, K.M.; DeCamp, D.; Roach, T.; Hsueh, R.; Rebres, R.A.; Ross, E.M.; Taussig, R.; et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem. 2007, 282, 10576–10584. [Google Scholar] [CrossRef] [Green Version]
- Canals, M.; Lane, J.R.; Wen, A.; Scammells, P.J.; Sexton, P.M.; Christopoulos, A. A Monod-Wyman-Changeux mechanism can explain G protein-coupled receptor (GPCR) allosteric modulation. J. Biol. Chem. 2012, 287, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, J.B.; Markey, S.P.; Ziffer, H. Preparation of 2(R) and 2(S) methyl-2-methylglycerates. Tetrahedron Asymmetry 1993, 4, 101–108. [Google Scholar] [CrossRef]
- Xu, X.; Feng, S.; Zhu, Y.; Li, H.; Shen, X.; Zhang, C.; Bai, J.; Zhang, L. Stereospecific radical polymerization of optically active (S)-N-(2-hydroxy-1-phenylethyl) methacrylamide catalyzed by Lewis acids. Eur. Polym. J. 2013, 49, 3673–3680. [Google Scholar] [CrossRef]
- Motulsky, H.J.C.; Christopoulos, A. Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting; GraphPad Software, Inc.: San Diego, CA, USA, 2003. [Google Scholar]
- Keov, P.; López, L.; Devine, S.M.; Valant, C.; Lane, J.R.; Scammells, P.J.; Sexton, P.M.; Christopoulos, A. Molecular mechanisms of bitopic ligand engagement with the M(1) muscarinic acetylcholine receptor. J. Biol. Chem. 2014, 289, 23817–23837. [Google Scholar] [CrossRef] [Green Version]
- Black, J.W.; Leff, P.; Shankley, N.P.; Wood, J. An operational model of pharmacological agonism: The effect of E/[A] curve shape on agonist dissociation constant estimation. Br. J. Pharmacol. 1985, 84, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Sibley, D.R.; Creese, I. Regulation of ligand binding to pituitary D-2 dopaminergic receptors. Effects of divalent cations and functional group modification. J. Biol. Chem. 1983, 258, 4957–4965. [Google Scholar] [CrossRef]
- Christopoulos, A. Assessing the distribution of parameters in models of ligand-receptor interaction: To log or not to log. Trends Pharmacol. Sci. 1998, 19, 351–357. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CPD | e.e. (%) | pKB (KB, µM) 1 | LogτB (τB) 2 | Logαβ (αβ) 3 |
|---|---|---|---|---|
| rac-2 | 0 | 5.80 ± 0.10 (1.6) | 0.32 ± 0.06 (2.1) | 2.00 ± 0.12 (100) |
| (S)-2 | 90 | 5.99 ± 0.09 (1.0) | 0.40 ± 0.06 (2.5) | 2.10 ± 0.25 (125) |
| (R)-2 | 84 | 5.12 ± 0.06 (7.6) *,^ | 0.46 ± 0.08 (2.9) | 1.51 ± 0.14 (31) *,^ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fyfe, T.J.; Scammells, P.J.; Lane, J.R.; Capuano, B. Enantioenriched Positive Allosteric Modulators Display Distinct Pharmacology at the Dopamine D1 Receptor. Molecules 2021, 26, 3799. https://doi.org/10.3390/molecules26133799
Fyfe TJ, Scammells PJ, Lane JR, Capuano B. Enantioenriched Positive Allosteric Modulators Display Distinct Pharmacology at the Dopamine D1 Receptor. Molecules. 2021; 26(13):3799. https://doi.org/10.3390/molecules26133799
Chicago/Turabian StyleFyfe, Tim J., Peter J. Scammells, J. Robert Lane, and Ben Capuano. 2021. "Enantioenriched Positive Allosteric Modulators Display Distinct Pharmacology at the Dopamine D1 Receptor" Molecules 26, no. 13: 3799. https://doi.org/10.3390/molecules26133799
APA StyleFyfe, T. J., Scammells, P. J., Lane, J. R., & Capuano, B. (2021). Enantioenriched Positive Allosteric Modulators Display Distinct Pharmacology at the Dopamine D1 Receptor. Molecules, 26(13), 3799. https://doi.org/10.3390/molecules26133799