
Organocatalytic Enantiospecific Total Synthesis of Butenolides

Abstract
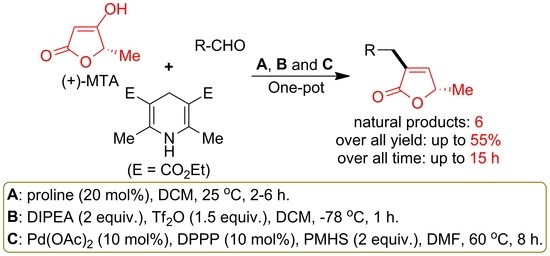
:
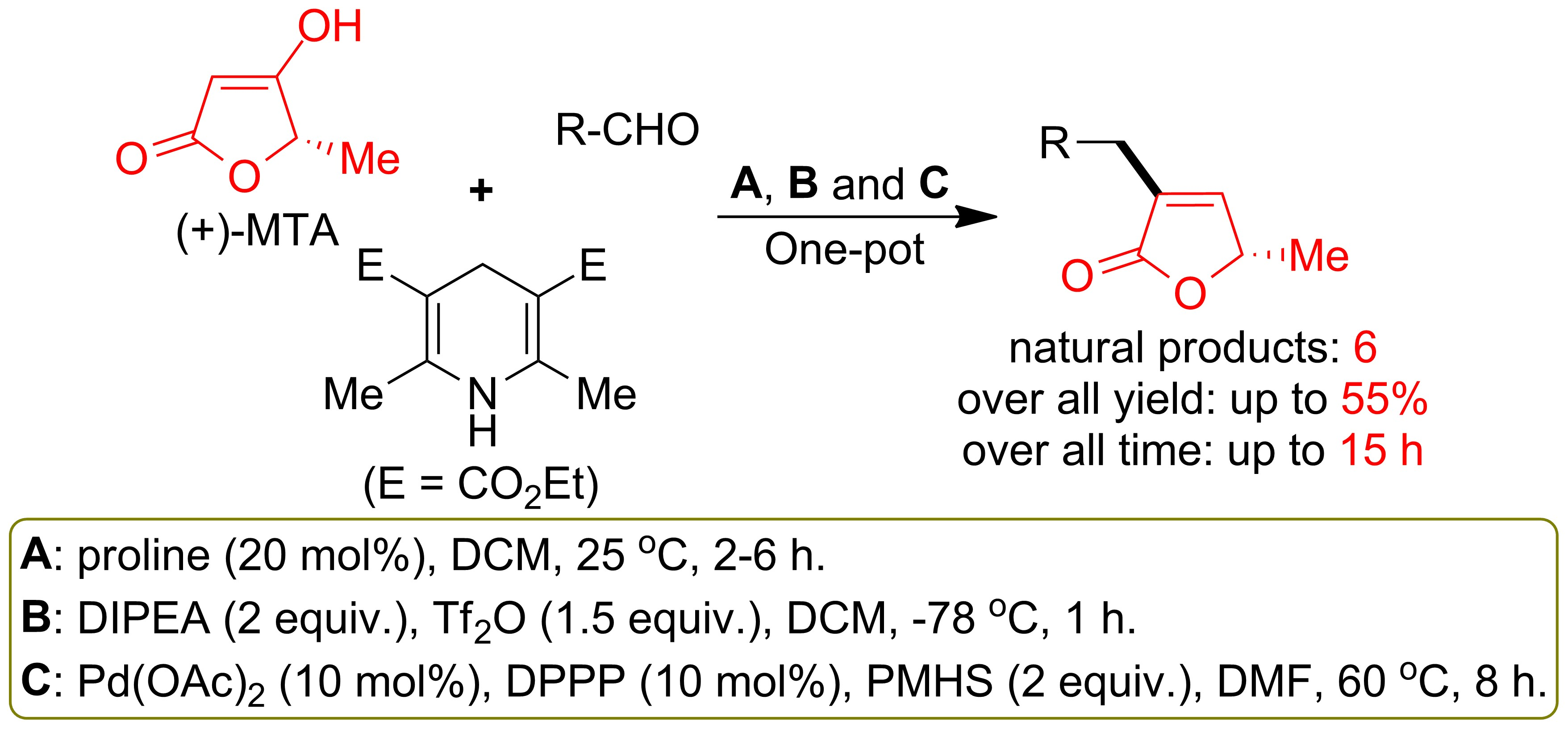{kind=link}
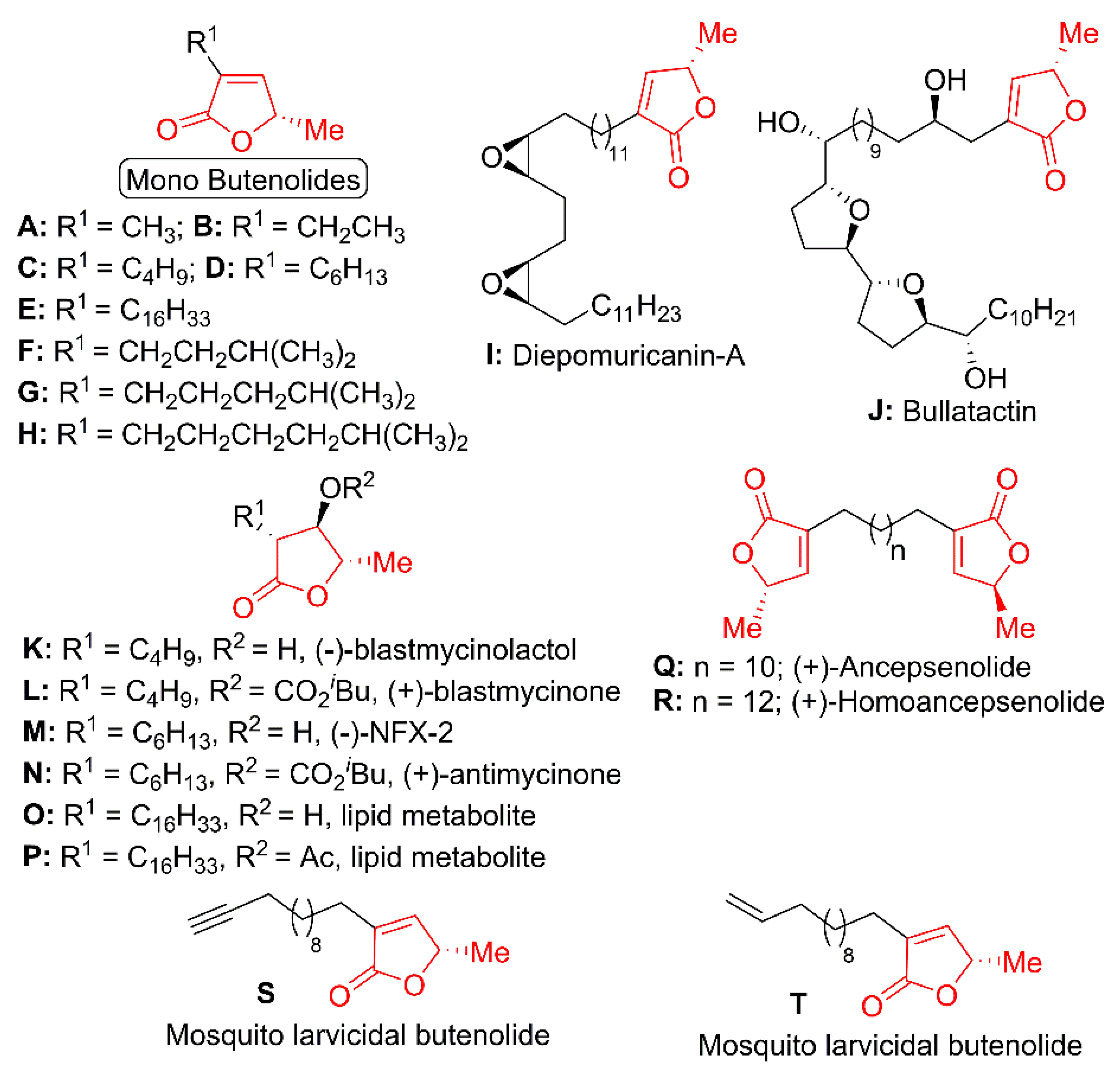{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
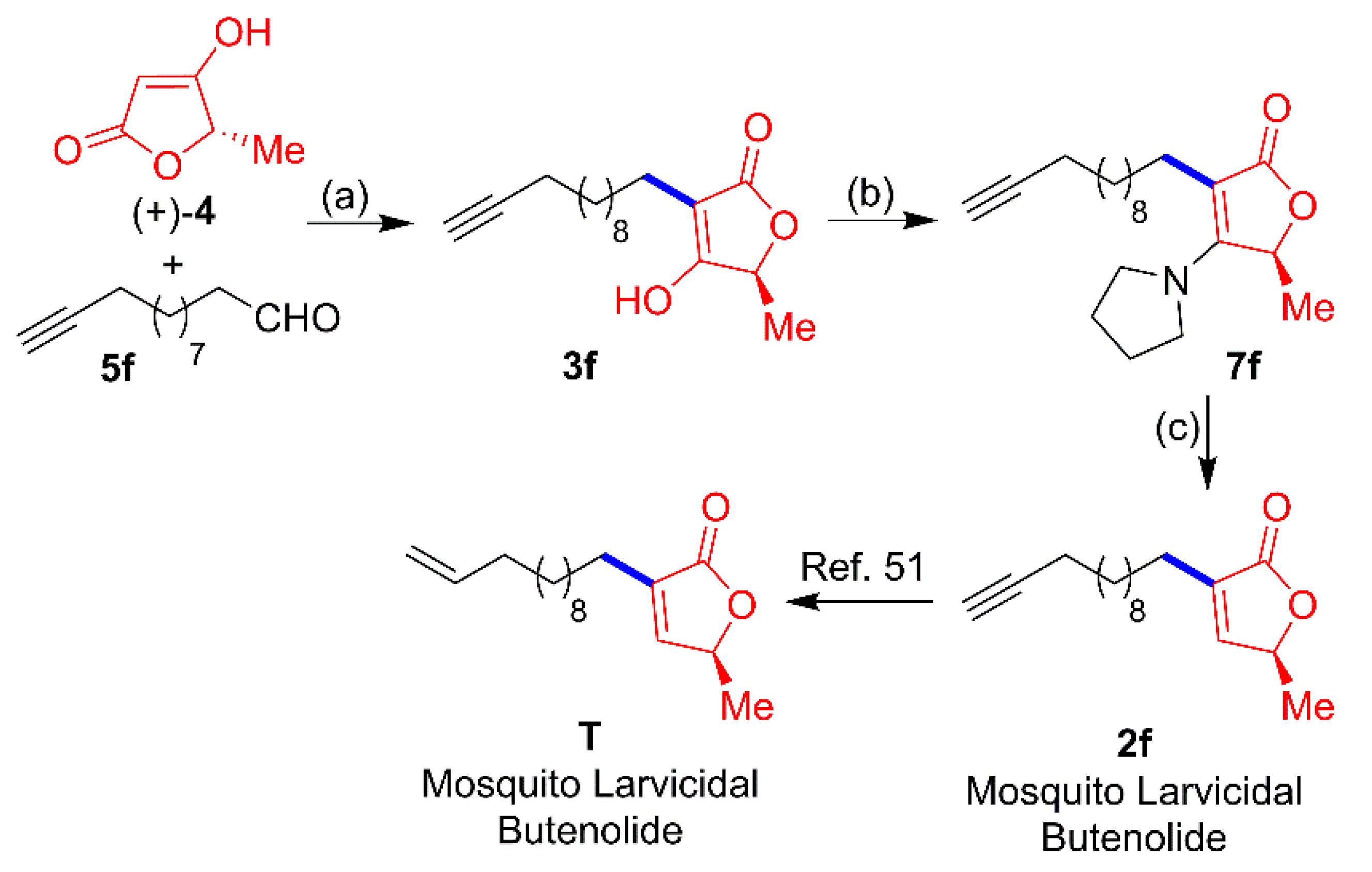{kind=link}
1. Introduction
2. Results and Discussion
3. Experimental Section
3.1. General Information
3.2. Materials
3.3. General Procedure for the Synthesis of OrgRC Products 3
3.4. General Procedure for the Synthesis of Products 2 through OrgRC and Palladium-Mediated Reductive Deoxygenation
3.5. General Procedure for the Synthesis of Products 2 through Silica-Mediated Reductive Deamination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Walji, A.; MacMillan, D.W.C. Strategies to bypass the taxol problem. enantioselective cascade catalysis, a new approach for the efficient construction of molecular complexity. Synlett 2007, 10, 1477–1489. [Google Scholar] [CrossRef]
- Jones, S.B.; Simmons, B.; Mastracchio, A.; MacMillan, D.W.C. Collective synthesis of natural products by means of organocascade catalysis. Nature 2011, 475, 183–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachary, D.B.; Jain, S. Sequential one-pot combination of multi-component and multi-catalysis cascade reactions: An emerging technology in organic synthesis. Org. Biomol. Chem. 2011, 9, 1277–1300. [Google Scholar] [CrossRef] [PubMed]
- Tietze, L.F. Domino reactions in organic synthesis. Chem. Rev. 1996, 96, 115–136. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Montagnon, T.; Snyder, S.A. Tandem reactions, cascade sequences, and biomimetic strategies in total synthesis. Chem. Commun. 2003, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Ramon, D.J.; Yus, M. Asymmetric multicomponent reactions (AMCRs): The new frontier. Angew. Chem. Int. Ed. 2005, 44, 1602–1634. [Google Scholar] [CrossRef] [PubMed]
- Erkkilä, A.; Majander, I.; Pihko, P.M. Iminium catalysis. Chem. Rev. 2007, 107, 5416–5470. [Google Scholar] [CrossRef]
- Grondal, C.; Jeanty, M.; Enders, D. Organocatalytic cascade reactions as a new tool in total synthesis. Nat. Chem. 2010, 2, 167–178. [Google Scholar] [CrossRef]
- Ramachary, D.B.; Reddy, Y.V. Dienamine catalysis: An emerging technology in organic synthesis. Eur. J. Org. Chem. 2012, 865–887. [Google Scholar] [CrossRef]
- Ramachary, D.B.; Chowdari, N.S.; Barbas III, C.F. Organocatalytic asymmetric domino Knoevenagel/Diels–Alder reactions: A bioorganic approach to the diastereospecific and enantioselective construction of highly substituted spiro[5,5]undecane-1,5,9-triones. Angew. Chem. Int. Ed. 2003, 42, 4233–4237. [Google Scholar] [CrossRef]
- Yang, J.W.; Fonseca, M.T.H.; List, B. Catalytic asymmetric reductive michael cyclization. J. Am. Chem. Soc. 2005, 127, 15036–15037. [Google Scholar] [CrossRef]
- Jiang, X.; Tan, B.; Barbas III, C.F. Core-structure-motivated design of iminium–enolate organocascade reactions: Enantioselective syntheses of 5,6-dihydroindolizines. Angew. Chem. Int. Ed. 2013, 52, 9261–9265. [Google Scholar] [CrossRef]
- Enders, D.; Hüttl, M.R.M.; Grondal, C.; Raabe, G. Control of four stereocentres in a triple cascade organocatalytic reaction. Nature 2006, 441, 861–863. [Google Scholar] [CrossRef]
- Carlone, A.; Cabrera, S.; Marigo, M.; Jørgensen, K.A. A new approach for an organocatalytic multicomponent domino asymmetric reaction. Angew. Chem. Int. Ed. 2007, 46, 1101–1104. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Jia, Z.-J.; Yin, X.; Wu, L.; Chen, Y.-C. Asymmetric quadruple aminocatalytic domino reactions to fused carbocycles incorporating a spirooxindole motif. Org. Lett. 2010, 12, 2766–2769. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Haack, K.-L.; Ieawsuwan, W.; Sunden, H.; Blanco, M.; Schoepke, F.R. Nature-inspired cascade catalysis: Reaction control through substrate concentration-double vs. quadruple domino reactions. Chem. Commun. 2011, 47, 3828–3830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, H.; Suzuki, T.; Hayashi, Y. High-yielding synthesis of the anti-influenza neuramidase inhibitor (−)-oseltamivir by three “one-pot” operations. Angew. Chem. Int. Ed. 2009, 48, 1304–1307. [Google Scholar] [CrossRef] [PubMed]
- Ramachary, D.B.; Venkaiah, C.; Krishna, P.M. Discovery of 2-aminobuta-1,3-enynes in asymmetric organocascade catalysis: Construction of drug-like spirocyclic cyclohexanes having five to six contiguous stereocenters. Chem. Commun. 2012, 48, 2252–2254. [Google Scholar] [CrossRef] [PubMed]
- Ramachary, D.B.; Reddy, Y.V.; Banerjee, A.; Banerjee, S. Design, synthesis and biological evaluation of optically pure functionalized spiro[5,5]undecane-1,5,9-triones as HIV-1 inhibitors. Org. Biomol. Chem. 2011, 9, 7282–7286. [Google Scholar] [CrossRef]
- Mangion, I.K.; MacMillan, D.W.C. Total synthesis of brasoside and littoralisone. J. Am. Chem. Soc. 2005, 127, 3696–3697. [Google Scholar] [CrossRef] [Green Version]
- Nicolaou, K.C.; Wu, T.R.; Sarlah, D.; Shaw, D.M.; Rowcliffe, E.; Burton, D.R. Total synthesis, revised structure, and biological evaluation of biyouyanagin A and analogues thereof. J. Am. Chem. Soc. 2008, 130, 11114–11121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, K.L.; Weise, C.F.; Dickmeiss, G.; Morana, F.; Davis, R.L.; Jørgensen, K.A. Selective access to both diastereoisomers in an enantioselective intramolecular Michael reaction by using a single chiral organocatalyst and application in the formal total synthesis of (−)-epibatidine. Chem. Eur. J. 2012, 18, 11913–11918. [Google Scholar] [CrossRef]
- Northrup, N.A.B.; MacMillan, D.W.C. Two-step synthesis of carbohydrates by selective aldol reactions. Science 2004, 305, 1752–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, C.L.; List, B. Catalytic, asymmetric transannular aldolizations: Total synthesis of (+)-hirsutene. J. Am. Chem. Soc. 2008, 130, 6737–6739. [Google Scholar] [CrossRef] [PubMed]
- Horning, B.D.; MacMillan, D.W.C. Nine-step enantioselective total synthesis of (−)-vincorine. J. Am. Chem. Soc. 2013, 135, 6442–6445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolaou, K.C.; Reingruber, R.; Sarlah, D.; Brase, S. Enantioselective intramolecular Friedel − Crafts-type α-arylation of aldehydes. J. Am. Chem. Soc. 2009, 131, 2086–2087. [Google Scholar] [CrossRef]
- Ramachary, D.B.; Kishor, M.; Reddy, Y.V. Development of pharmaceutical drugs, drug intermediates and ingredients by using direct organo-click reactions. Eur. J. Org. Chem. 2008, 975–993. [Google Scholar] [CrossRef]
- Ramachary, D.B.; Kishor, M. Organocatalytic sequential one-pot double cascade asymmetric synthesis of Wieland − Miescher ketone analogues from a Knoevenagel/hydrogenation/Robinson annulation sequence: Scope and applications of organocatalytic biomimetic reductions. J. Org. Chem. 2007, 72, 5056–5068. [Google Scholar] [CrossRef] [PubMed]
- Ramachary, D.B.; Reddy, Y.V. A general approach to chiral building blocks via direct amino acid-catalyzed cascade three-component reductive alkylations: Formal total synthesis of HIV-1 protease inhibitors, antibiotic agglomerins, brefeldin A, and (R)-γ-hexanolide. J. Org. Chem. 2010, 75, 74–85. [Google Scholar] [CrossRef]
- Ide, N.D.; Ragan, J.A.; Bellavance, G.; Brenek, S.J.; Cordi, E.M.; Jensen, G.O.; Jones, K.N.; LaFrance, D.; Leeman, K.R.; Letendre, L.J.; et al. Synthesis of filibuvir. part III. development of a process for the reductive coupling of an aldehyde and a β-keto-lactone. Org. Process. Res. Dev. 2014, 18, 45–56. [Google Scholar] [CrossRef]
- Tummatorn, J.; Dudley, G.B. Generation of medium-ring cycloalkynes by ring expansion of vinylogous acyl triflates. Org. Lett. 2011, 13, 1572–1575. [Google Scholar] [CrossRef]
- Wong, Y.-C.; Tseng, C.-T.; Kao, T.-T.; Yeh, Y.-C.; Shia, K.-S. Tandem cyclization of α-cyano α-alkynyl aryl ketones induced by tert-butyl hydroperoxide and tetrabutylammonium iodide. Org. Lett. 2012, 14, 6024–6027. [Google Scholar] [CrossRef]
- Miao, L.; Shu, H.; Noble, A.R.; Fournet, S.P.; Stevens, E.D.; Trudell, M.L. An enantioselective formal synthesis of (+)-Gephyrotoxin 287C. Arkivoc 2010, 4, 6–14. [Google Scholar]
- Hiroya, K.; Suwa, Y.; Ichihashi, Y.; Inamoto, K.; Doi, T. Total synthesis of optically active lycopladine A by utilizing diastereoselective protection of carbonyl group in a 1,3-cyclohexanedione derivative. J. Org. Chem. 2011, 76, 4522–4532. [Google Scholar] [CrossRef]
- Elamparuthi, E.; Fellay, C.; Neuburger, M.; Gademann, K. Total synthesis of cyrneine A. Angew. Chem. Int. Ed. 2012, 51, 4071–4073. [Google Scholar] [CrossRef] [PubMed]
- Gerber, N.N. Volatile lactones from streptomyces. Tetrahedron Lett. 1973, 10, 771–774. [Google Scholar] [CrossRef]
- Rodrfguez, A.D.; Ramfrez, C. Further butenolides from the caribbean octocoral pterogorgia citrina. J. Nat. Prod. 1994, 57, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Corbera, J.; Font, J.; Monsalvatje, M.; Ortuiio, R.M.; Sanchez-Ferrando, F. Alkylation of (1S,2R,5R,6S,7R)- and (1R,2R,5R,6S,7S)-5-methyl-4-oxatricyclo[5.2.1.02,6]-8-decen-3-one. application to the synthesis of (R)-3-alkyl-5-methyl-2(5H)-furanones. J. Org. Chem. 1988, 53, 4393–4395. [Google Scholar] [CrossRef]
- Esposti, M.D.; Ghelli, A.; Ratta, M.; Cortes, D.; Estornell, E. Natural substances (acetogenins) from the family annonaceae are powerful inhibitors of mitochondrial NADH dehydrogenase (complex I). Biochem. J. 1994, 301, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratnayake, R.; Karunaratne, V.; Bandara, B.M.R.; Kumar, V.; MacLeod, J.K.; Simmonds, P. Two new lactones with mosquito larvicidal activity from three hortonia species. J. Nat. Prod. 2001, 64, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, M.; Brito, I.; Cueto, M.; D’Croz, L.; Darias, J. 13C NMR-Based empirical rules to determine the configuration of fatty acid butanolides. novel γ-dilactones from pterogorgia spp. Org. Lett. 2006, 8, 5001–5004. [Google Scholar] [CrossRef]
- Larson, G.L.; Betancourt de Perez, R.M. A two-step preparation of α-alkylidene γ-lactones from γ-lactones: A synthesis of (±)-ancepsenolide. J. Org. Chem. 1985, 50, 5257–5260. [Google Scholar] [CrossRef]
- Podraza, K.F.; Snede, A.T. A short synthesis of ancepsenolide. J. Nat. Prod. 1985, 48, 792–795. [Google Scholar] [CrossRef]
- Berkenbusch, T.; Bruckner, R. Brief syntheses of (+)-blastmycinone and related γ-lactones from an asymmetrically dihydroxylated carboxylic ester. Tetrahedron 1998, 54, 11461–11470. [Google Scholar] [CrossRef]
- Nishide, K.; Aramata, A.; Kamanaka, T.; Inoue, T.; Node, M. Total asymmetric syntheses of (+)-blastmycinone and related γ-lactones. Tetrahedron 1994, 50, 8337–8346. [Google Scholar] [CrossRef]
- Chen, M.-J.; Lo, C.-Y.; Chin, C.-C.; Liu, R.-S. Total synthesis of (+)-blastmycinone, (−)-litsenolide C1, and related natural trisubstituted lactones via alkynyltungsten compounds. J. Org. Chem. 2000, 65, 6362–6367. [Google Scholar] [CrossRef] [PubMed]
- Pashkovsky, F.S.; Katok, Y.M.; Khlebnicova, T.S.; Lakhvich, F.A. A new efficient synthesis of alkyl substituted Δ2-butenolides. Tetrahedron Lett. 2001, 42, 3657–3658. [Google Scholar] [CrossRef]
- Ghobril, C.; Kister, J.; Baati, R. A synthetic route to α-substituted butenolides: Enantioselective synthesis of (+)-ancepsenolide. Eur. J. Org. Chem. 2011, 3416–3419. [Google Scholar] [CrossRef]
- Ferrarini, R.S.; Dos Santos, A.A.; Comasseto, J.V. Tellurium in organic synthesis: A new approach to trisubstituted γ-butyrolactones with trans–trans relative stereochemistry. Total enantioselective synthesis of (−)-blastmycinolactol, (+)-blastmycinone, (−)-NFX-2, and (+)-antimycinone. Tetrahedron Lett. 2010, 51, 6843–6846. [Google Scholar] [CrossRef]
- Pace, V.; Rae, J.P.; Harb, H.Y.; Procter, D.J. NHC–Cu(i) catalysed asymmetric conjugate silyl transfer to unsaturated lactones: Application in kinetic resolution. Chem. Commun. 2013, 49, 5150–5152. [Google Scholar] [CrossRef] [Green Version]
- He, Y.-T.; Yang, H.-N.; Yao, Z.-J. Sharpless AD strategy towards the γ-methyl butenolide unit of acetogenins: Enantioselective synthesis of butenolide I and II with mosquito larvicidal activity. Tetrahedron 2002, 58, 8805–8810. [Google Scholar] [CrossRef]
- Bernardi, A.; Beretta, M.G.; Colombo, L.; Gennari, C.; Poli, G.; Scolastico, C. Synthetic opportunities offered by anti α-methylene-/3-hydroxy-7-aIkoxy esters: Stereoselective reactions at the double bond. J. Org. Chem. 1985, 50, 4442–4447. [Google Scholar] [CrossRef]
- Tsunoda, T.; Nishii, T.; Yoshizuka, M.; Yamasaki, C.; Suzuki, T.; Ito, S. A total synthesis of (−)-antimycin A3b. Tetrahedron Lett. 2000, 41, 7667–7671. [Google Scholar] [CrossRef]
- Amonkar, C.P.; Tilve, S.G.; Parameswaran, P.S. Convenient synthesis of volatile streptomyces lactones. Synthesis 2005, 2341–2344. [Google Scholar] [CrossRef]
- Ortuno, R.M.; Bigorra, J.; Font, J. Stereocontrolled synthesis of (3S,4S,5S)-3-alkyl-4-hydroxy-5-methyl-2(3H)-dihydrofubanones and derivatives: Configurational assignment of some clinostemon mahuba and plexaura flava metabolites. Tetrahedron 1988, 44, 5139–5144. [Google Scholar] [CrossRef]
- Trost, B.M.; Muller, T.J.J. Butenolide synthesis based upon a contra-electronic addition in a ruthenium-catalyzed Alder ene reaction. synthesis and absolute configuration of (+)-ancepsenolide. J. Am. Chem. Soc. 1994, 116, 4985–4986. [Google Scholar] [CrossRef]
- Trost, B.M.; Muller, T.J.J.; Martinez, J. Ruthenium-catalyzed synthesis of butenolides and pentenolides via contra-electronic α-alkylation of hydroxyalkynoates. J. Am. Chem. Soc. 1995, 117, 1888–1899. [Google Scholar] [CrossRef]
- Takai, K.; Iriye, R. Enantioselective synthesis of ancepsenolide and its analogs. Biosci. Biotechnol. Biochem. 2001, 65, 1903–1906. [Google Scholar] [CrossRef]
- Jiang, S.; Wu, Y.-L.; Yao, Z.-J. First synthesis of mosquito larvicidal butenolides I and II. Chin. J. Chem. 2002, 20, 692–696. [Google Scholar] [CrossRef]
- Gharpure, S.J.; Nanda, L.N.; Shukla, M.K. Donor–acceptor substituted cyclopropane to butanolide and butenolide natural products: Enantiospecific first total synthesis of (+)-hydroxyancepsenolide. Org. Lett. 2014, 16, 6424–6427. [Google Scholar] [CrossRef] [PubMed]
- Brandange, S.; Flodman, L.; Norberg, A. Studies on the intramolecular Claisen condensation; facile synthesis of tetronic acids. J. Org. Chem. 1984, 49, 927–928. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madhavachary, R.; Mallik, R.; Ramachary, D.B. Organocatalytic Enantiospecific Total Synthesis of Butenolides. Molecules 2021, 26, 4320. https://doi.org/10.3390/molecules26144320
Madhavachary R, Mallik R, Ramachary DB. Organocatalytic Enantiospecific Total Synthesis of Butenolides. Molecules. 2021; 26(14):4320. https://doi.org/10.3390/molecules26144320
Chicago/Turabian StyleMadhavachary, Rudrakshula, Rosy Mallik, and Dhevalapally B. Ramachary. 2021. "Organocatalytic Enantiospecific Total Synthesis of Butenolides" Molecules 26, no. 14: 4320. https://doi.org/10.3390/molecules26144320
APA StyleMadhavachary, R., Mallik, R., & Ramachary, D. B. (2021). Organocatalytic Enantiospecific Total Synthesis of Butenolides. Molecules, 26(14), 4320. https://doi.org/10.3390/molecules26144320