
Base-Mediated Claisen Rearrangement of CF3-Containing Bisallyl Ethers

Abstract
:
1. Introduction
2. Results and Discussion
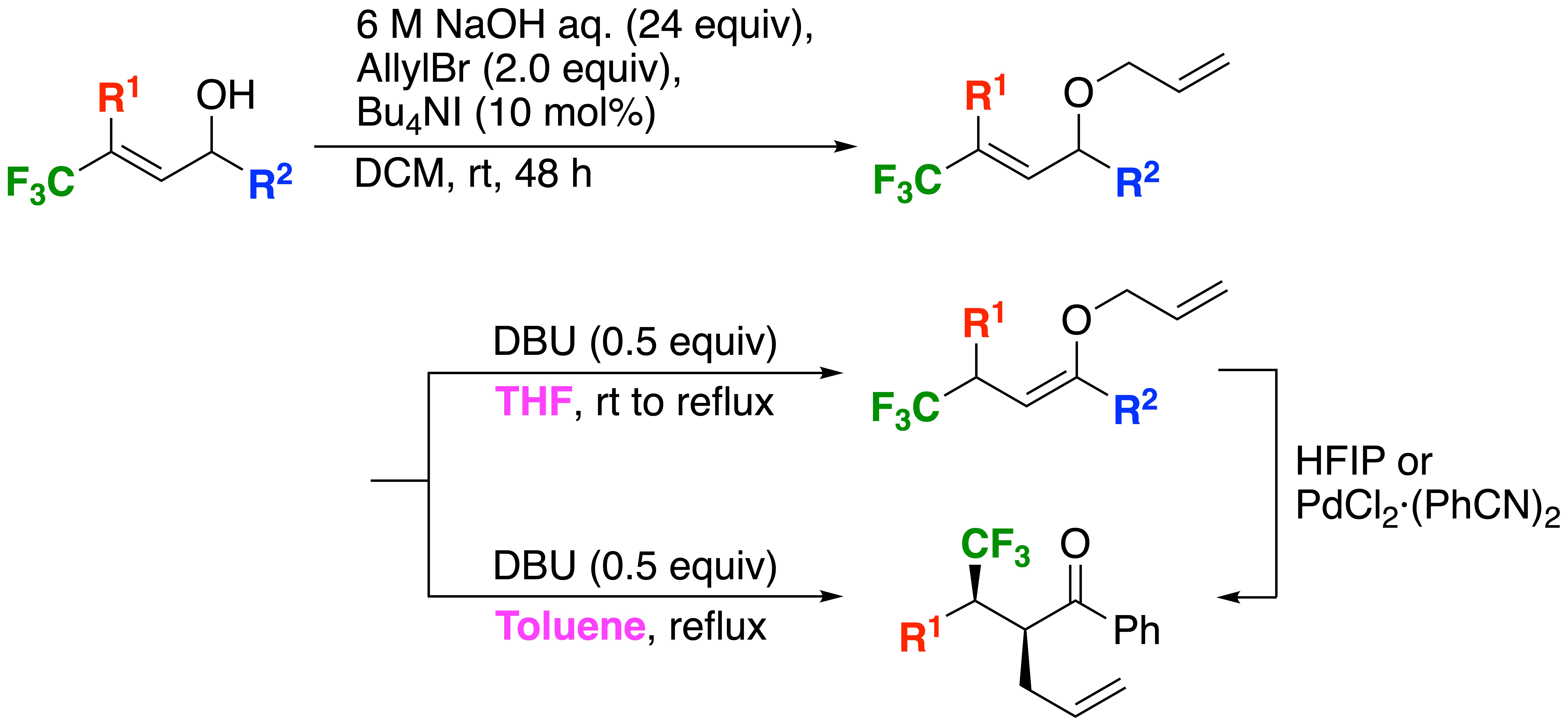
2.1. Preparation of Bisallyl Ethers 3
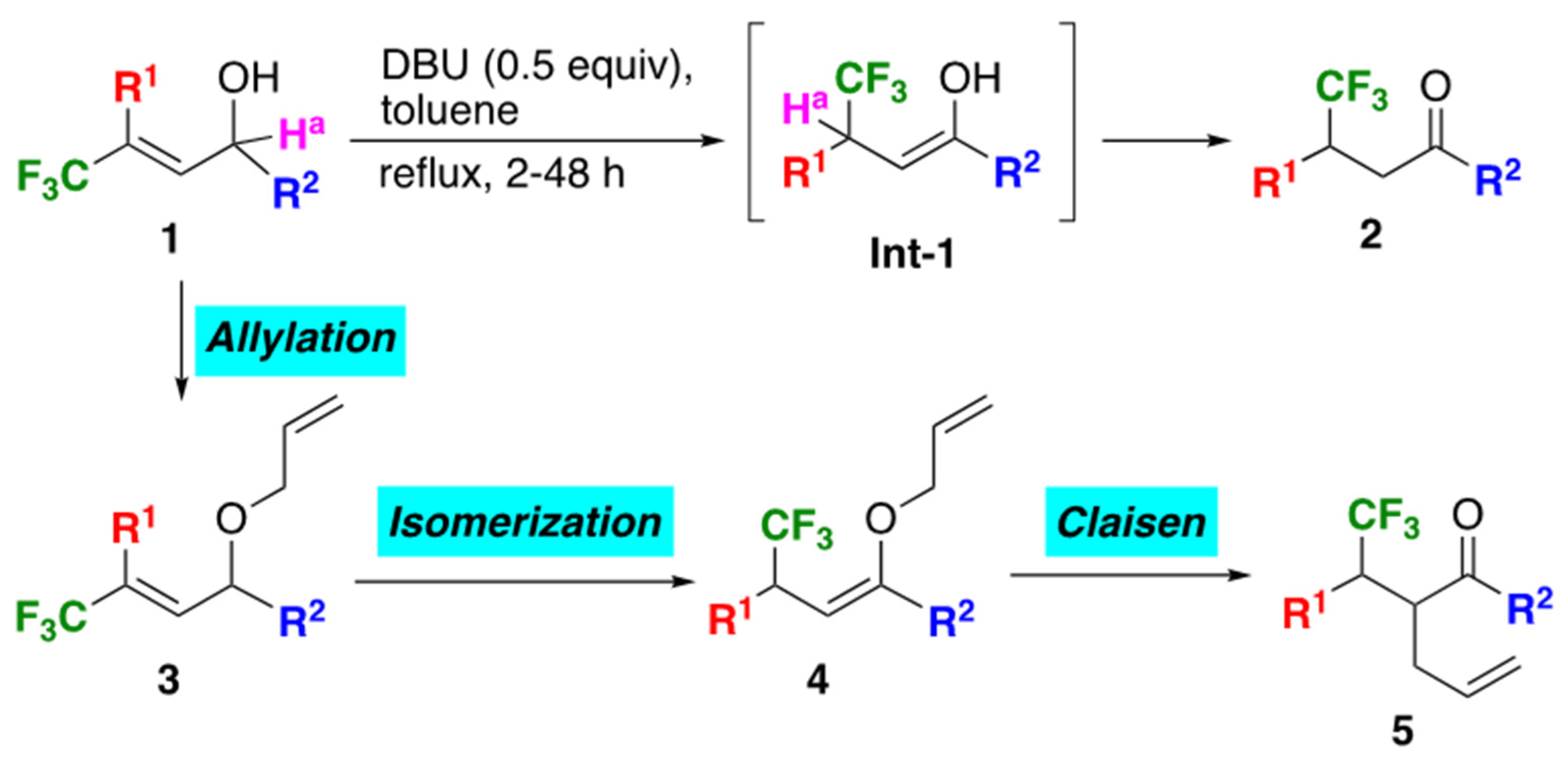
2.2. Preparation of Allyl Vinyl Ethers 4 and One-Pot Isomerization-Claisen Rearrangement from 3
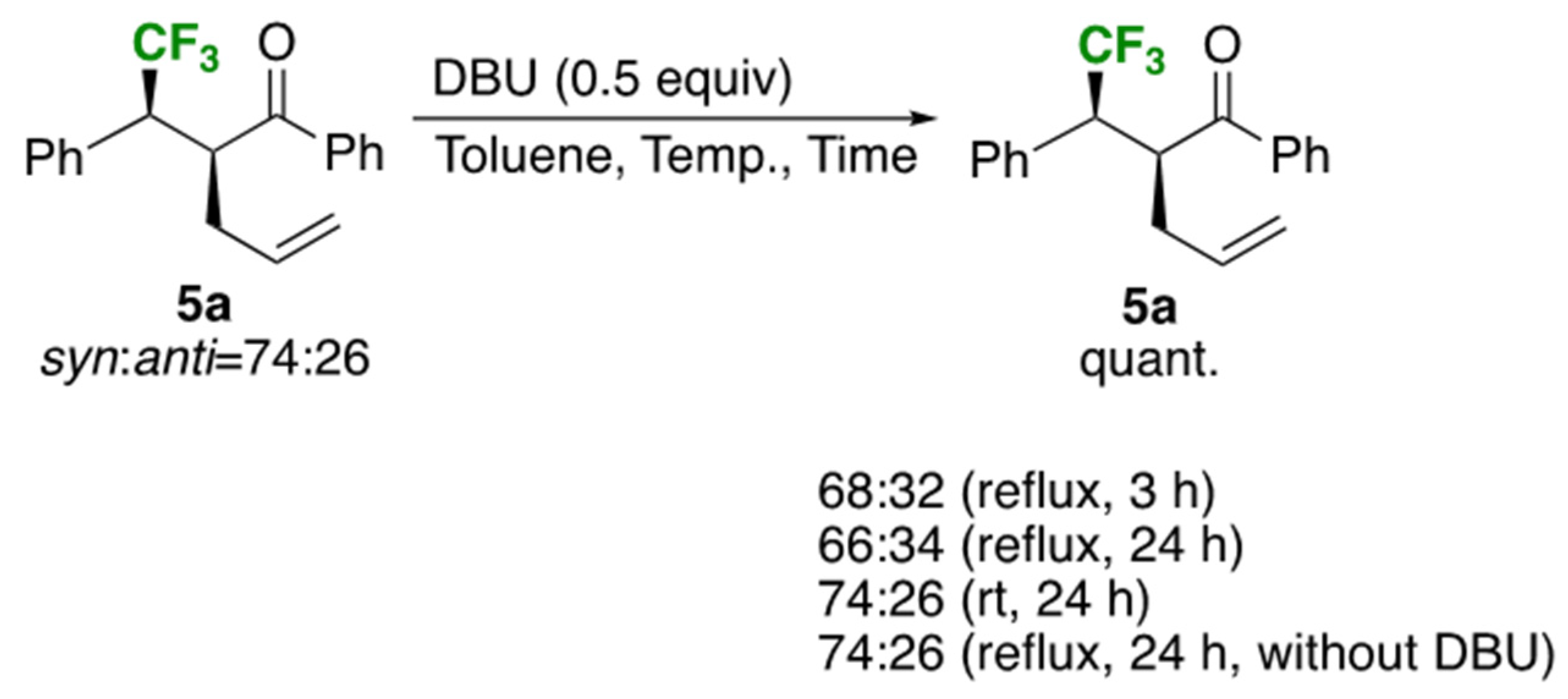
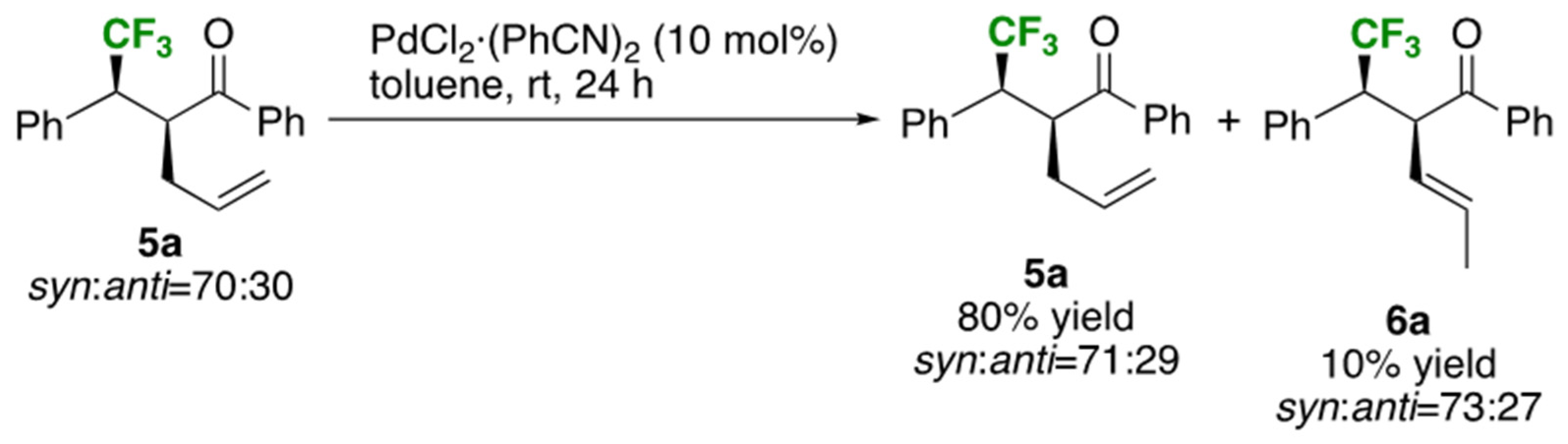
2.3. Improvement of the Diastereoselectivity of the Claisen Rearrangement Products 5
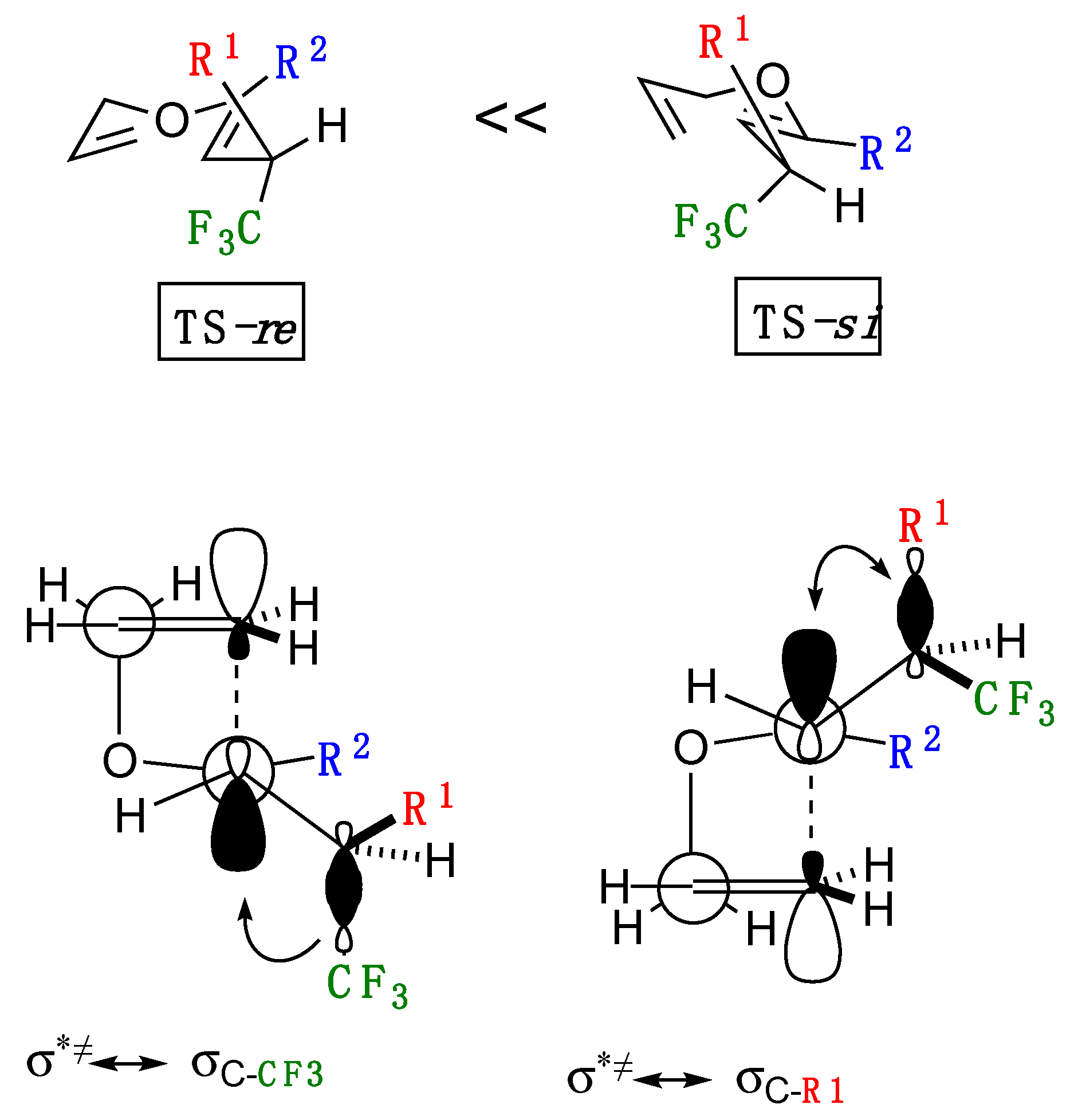
2.4. Discussion on the Reaction Mechanism
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. General Procedure for the Preparation of Bisallyl Ethers
4.2.1. (E)-1,1,1-Trifluoro-2,4-diphenyl-4-{(prop-2-en-1-yl)oxy}but-2-ene (3a)
4.2.2. (E)-1,1,1-Trifluoro-2-(4-methoxyphenyl)-4-phenyl-4-{(prop-2-en-1-yl)oxy}but-2-ene (3b)
4.2.3. (E)-1,1,1-Trifluoro-2-(4-fluorophenyl)-4-phenyl-4-{(prop-2-en-1-yl)oxy}but-2-ene (3c)
4.2.4. (E)-1-Phenyl-1-{(prop-2-en-1-yl)oxy}-3-(trifluoromethyl)pent-2-ene (3d)
4.2.5. (E)-1,5-Diphenyl-1-{(prop-2-en-1-yl)oxy}-3-(trifluoromethyl)pent-2-ene (3e)
4.2.6. (E)-1-(4-Methoxyphenyl)-5-phenyl-1-{(prop-2-en-1-yl)oxy}-3-(trifluoromethyl)-pent-2-ene (3f)
4.2.7. (E)-1-(4-Bromophenyl)-5-phenyl-1-{(prop-2-en-1-yl)oxy}-3-(trifluoromethyl)pent-2-ene (3g)
4.3. General Procedure for the Preparation of Ally Vinyl Ethers
4.3.1. (E)-4,4,4-Trifluoro-1,3-diphenyl-1-{(prop-2-en-1-yl)oxy}but-1-ene (4a)
4.3.2. (E)-4,4,4-Trifluoro-3-(4-methoxyphenyl)-1-phenyl-1-{(prop-2-en-1-yl)oxy}but-1-ene (4b)
4.3.3. (E)-4,4,4-Trifluoro-3-(4-fluorophenyl)-1-phenyl-1-{(prop-2-en-1-yl)oxy}but-1-ene (4c)
4.3.4. (E)-1-Phenyl-1-{(prop-2-en-1-yl)oxy}-3-(trifluoromethyl)pent-1-ene (4d)
4.3.5. (E)-1,5-Diphenyl-1-{(prop-2-en-1-yl)oxy}-3-(trifluoromethyl)pent-1-ene (4e)
4.3.6. (E)-1-(4-Methoxyphenyl)-5-phenyl-1-{(prop-2-en-1-yl)oxy}-3-(trifluoromethyl)-pent-1-ene (4f)
4.3.7. (E)-1-(4-Bromophenyl)-5-phenyl-1-{(prop-2-en-1-yl)oxy}-3-(trifluoromethyl)pent-1-ene (4g)
4.4. General Procedure for the Claisen Rearrangement of Ally Vinyl Ethers
4.4.1. 4,4,4-Trifluoro-1,3-diphenyl-2-(prop-2-en-1-yl)butan-1-one (5a)
Method 1. By Heating (Isomerization-Rearrangement)
Method 2. Rearrangement of Enol Ethers with the Aid of a Palladium Catalyst
Major Isomer
Minor Isomer
4.4.2. 4,4,4-Trifluoro-3-(4-methoxyphenyl)-1-phenyl-2-(prop-2-en-1-yl)butan-1-one (5b)
Method 1
Method 2
Major Isomer
Minor Isomer
4.4.3. 4,4,4-Trifluoro-3-(4-fluorophenyl)-1-phenyl-2- (prop-2-en-1-yl)butan-1-one (5c)
Method 1
Method 2
Major Isomer
Minor Isomer
4.4.4. 1-Phenyl-2-(prop-2-en-1-yl)-3-(trifluoromethyl)pentan-1-one (5d)
Method 1
Method 2
4.4.5. 1,5-Diphenyl-2-(prop-2-en-1-yl)-3-(trifluoromethyl)pentan-1-one (5e)
Method 1
4.4.6. 1-(4-Methoxyphenyl)-5-phenyl-2-(prop-2-en-1-yl)-3-(trifluoromethyl)pentan-1-one (5f)
Method 1
4.4.7. 1-(4-Bromophenyl)-5-phenyl-2-(prop-2-en-1-yl)-3-(trifluoromethyl)pentan-1-one (5g)
Method 1
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- O’Hagan, D. Polar Organofluorine Substituents: Multivicinal Fluorines on Alkyl Chains and Alicyclic Rings. Chem. Eur. J. 2020, 26, 7981–7997. [Google Scholar] [CrossRef]
- Brittain, W.D.; Lloyd, C.M.; Cobb, S.L. Synthesis of Complex Unnatural Fluorine-containing Amino Acids. J. Fluor. Chem. 2020, 239, 109630. [Google Scholar] [CrossRef] [PubMed]
- Remete, A.M.; Kiss, L. Synthesis of Fluorine-Containing Molecular Entities Through Fluoride Ring Opening of Oxiranes and Aziridines. Eur. J. Org. Chem. 2019, 2019, 5574–5602. [Google Scholar] [CrossRef] [Green Version]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef]
- Orsi, D.L.; Altman, R.A. Exploiting the Unusual Effects of Fluorine in Methodology. Chem. Commun. 2017, 53, 7168–7181. [Google Scholar] [CrossRef]
- Yamazaki, T.; Kawasaki-Takasuka, T.; Furuta, A.; Sakamoto, S. Facile Conversion of 4,4,4-Trifluorobut-2-yn-1-ols to 4,4,4-Trifluorobut-2-en-1-ones. Tetrahedron 2009, 65, 5945–5948. [Google Scholar] [CrossRef]
- Hamada, Y.; Kawasaki-Takasuka, T.; Yamazaki, T. Base-promoted Isomerization of CF3-containing Allylic Alcohols to the Corresponding Saturated Ketones under Metal-free Conditions. Beilstein J. Org. Chem. 2017, 13, 1507–1512. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Erro, S.; Sanz-Marco, A.; Gómez, A.B.; Vázquez-Romero, A.; Ahlquist, M.S.G.; Martín-Matute, B. Base-Catalyzed Stereospecific Isomerization of Electron-Deficient Allylic Alcohols and Ethers through Ion-Pairing. J. Am. Chem. Soc. 2016, 138, 13408–13414. [Google Scholar] [CrossRef]
- Yamazaki, T.; Shinohara, N.; Kitazume, T.; Sato, S. Highly Diastereoselective Sequential Enolate-Michael Addition-Ireland Claisen Rearrangement. J. Org. Chem. 1995, 60, 8140–8141. [Google Scholar] [CrossRef]
- Lu, Y.; Goldstein, E.; Stoltz, B.M. Palladium-Catalyzed Enantioselective Csp3–Csp3 Cross-Coupling for the Synthesis of (Poly)fluorinated Chiral Building Blocks. Org. Lett. 2018, 20, 5657–5660. [Google Scholar] [CrossRef] [Green Version]
- Alexy, E.J.; Zhang, H.; Stoltz, B.M. Catalytic Enantioselective Synthesis of Acyclic Quaternary Centers: Palladium-Catalyzed Decarboxylative Allylic Alkylation of Fully Substituted Acyclic Enol Carbonates. J. Am. Chem. Soc. 2018, 140, 10109–10112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czekelius, C.; Erdbrink, H. Stereoselective Synthesis of Fluoroalkylated Butanolides. Synlett 2013, 24, 2383–2388. [Google Scholar] [CrossRef] [Green Version]
- Canney, D.J.; Lu, H.-F.; McKeon, A.C.; Yoon, K.-W.; Xu, K.; Holland, K.D.; Rothman, S.M.; Ferrendelli, J.A.; Covey, D.F. Structure-activity Studies of Fluoroalkyl-substituted γ-Butyrolactone and γ-Thiobutyrolactone Modulators of GABAA Receptor Function. Bioorg. Med. Chem. 1998, 6, 43–55. [Google Scholar] [CrossRef]
- Milburn, R.R.; McRae, K.; Chan, J.; Tedrow, J.; Larsen, R.; Faul, M. A Practical Preparation of Aryl β-Ketophosphonates. Tetrahedron Lett. 2009, 50, 870–872. [Google Scholar] [CrossRef]
- Yamazaki, T.; Mano, N.; Hikage, R.; Kaneko, T.; Kawasaki-Takasuka, T.; Yamada, S. Convenient Stereoselective Synthesis of β-Perfluoroalkyl α,β-Unsaturated Esters via Horner–Wadsworth–Emmons Reactions. Tetrahedron 2015, 71, 8059–8066. [Google Scholar] [CrossRef]
- Evans, D.A.; Nelson, J.V.; Taber, T.R. Stereoselective Aldol Condensations. In Topic in Stereochemistry; Allinger, N.L., Eliel, E.L., Wilen, S.H., Eds.; Wiley: New York, NY, USA, 1982; Volume 13, pp. 1–115. [Google Scholar]
- Hiersemann, M.; Rehbein, J. Claisen Rearrangement of Aliphatic Allyl Vinyl Ethers from 1912 to 2012: 100 Years of Electrophilic Catalysis. Synthesis 2013, 45, 1121–1159. [Google Scholar] [CrossRef]
- Bonnet-Delpon, D.; Bégué, J.-P.; Crousse, B. Fluorinated Alcohols: A New Medium for Selective and Clean Reaction. Synlett 2003, 2004, 18–29. [Google Scholar] [CrossRef]
- Ziegler, F.E. The Thermal, Aliphatic Claisen Rearrangement. Chem. Rev. 1988, 88, 1423–1452. [Google Scholar] [CrossRef]
- Hiersemann, M.; Abraham, L. Catalysis of the Claisen Rearrangement of Aliphatic Allyl Vinyl Ethers. Eur. J. Org. Chem. 2002, 1461–1471. [Google Scholar] [CrossRef]
- van der Baan, J.; Bickelhaupt, F. Palladium(II)-catalyzed Claisen Rearrangement of Allyl Vinyl Ethers. Tetrahedron Lett. 1986, 27, 6267–6270. [Google Scholar] [CrossRef]
- Yamazaki, T.; Haga, J.; Kitazume, T. Stereoselective Michael Addition Reactions of Acylated Oxazolidinones to Ethyl 3-Trifluoromethylacrylate. Chem. Lett. 1991, 20, 2175–2178. [Google Scholar] [CrossRef]
- Yamazaki, T.; Haga, J.; Kitazume, T.; Nakamura, S. Highly Diastereoselective Michael Addition Reactions of Lithium Enolates to Ethyl 3-Trifluoromethylacrylate. Chem. Lett. 1991, 20, 2171–2174. [Google Scholar] [CrossRef]
- Yamazaki, T.; Ichige, T.; Takei, S.; Kawashita, S.; Kitazume, T.; Kubota, T. Effect of Allylic CH3-nFnGroups (n= 1−3) on π-Facial Diastereoselection. Org. Lett. 2001, 3, 2915–2918. [Google Scholar] [CrossRef]
- Cieplak, A.S. Inductive and Resonance Effects of Substituents on π-Face Selection. Chem. Rev. 1999, 99, 1265–1336. [Google Scholar] [CrossRef] [PubMed]
- MacPhee, J.A.; Panaye, A.; Dubois, J.-E. Steric Effects—I. A Critical Examination of the Taft Steric Parameters–Es. Definition of a Revised, Broader and Homogeneous Scale. Extension to Highly Congested Alkyl Groups. Tetrahedron 1978, 34, 3553–3562. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Base | AllylBr | Time | Isolated Yield 1 (%) | Recovery 1 | ||||
|---|---|---|---|---|---|---|---|---|
| Entry | (equiv) | PTC | Solvent 2 | (equiv) | (h) | 3a | 2a | (%) |
| 1 | NaH (1.1) | THF | 1.1 | 16 | (4) | 35 | (0) | |
| 2 | t-BuOK (2.0) | THF | 2.0 | 16 | (45) | 17 | (35) | |
| 3 | DBU (1.1) | THF | 5.0 | 16 | (0) | (0) | (99) | |
| 4 | BuLi (1.1) | THF | 1.1 | 16 | (0) | (0) | (99) | |
| 5 | NaOH 3 (24) | Bu4NI | DCM | 5.0 | 16 | 92 | trace | (0) |
| 6 | NaOH 3 (24) | BnEt3NCl | DCM | 5.0 | 16 | (79) | (2) | (18) |
| 7 | NaOH 3 (24) | Bu4NBr | DCM | 5.0 | 16 | 92 | (2) | (0) |
| 8 | NaOH 3 (24) | Bu4NI | THF | 5.0 | 16 | (82) | trace | (18) |
| 9 | NaOH 3 (24) | Bu4NI | CHCl3 | 5.0 | 16 | 41 | (0) | (0) |
| 10 | NaOH 3 (24) | Bu4NI | DCM | 4.0 | 16 | 92 | trace | Trace |
| 11 | NaOH 3 (24) | Bu4NI | DCM | 3.0 | 16 | 88 | (2) | (0) |
| 12 | NaOH 3 (24) | Bu4NI | DCM | 2.0 | 16 | 89 | (3) | trace |
| 13 | NaOH 3 (24) | Bu4NI | DCM | 1.5 | 24 | (79) | trace | trace |
| 14 | NaOH 3 (24) | Bu4NI | DCM | 1.1 | 48 | 74 | (6) | (0) |
| 15 | BuLi 5,6 (1.1) | THF | 1.1 | 16 | (24) | (15) | (60) | |
| 16 | BuLi 5,7 (1.0) | THF | 1.0 | 6 | (58) | (0) | (42) | |
| 17 | NaOH 4 (24) | Bu4NI | DCM | 2.0 | 16 | (54) | (2) | (44) |
| 18 | NaOH 4 (24) | Bu4NI | DCM | 2.0 | 16 | (69) | (6) | (25) |
| 19 | NaOH 3 (12) | Bu4NI | DCM | 2.0 | 16 | (77) | (3) | (16) |
| 20 | NaOH 3 (6) | Bu4NI | DCM | 2.0 | 16 | (39) | (2) | (58) |
| 21 | NaOH 3 (24) | Bu4NI | DCM | 2.0 | 16 | 69 | (2) | (29) |
| 22 | NaOH 3 (24) | Bu4NI | DCM | 2.0 | 24 | 83 | (4) | (13) |
| 23 | NaOH 3 (24) | Bu4NI | DCM | 2.0 | 48 | 98 | trace | trace |

| Isolated Yield 1 (%) | |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Comp. | 3 | 2 |
| 1 | Ph | Ph | a | 98 | (2) |
| 2 | p-MeOC6H4 | Ph | b | 90 | (2) |
| 3 | p-FC6H4 | Ph | c | 84 | (7) |
| 4 | Et | Ph | d | 90 | (trace) |
| 5 | Ph(CH2)2 | Ph | e | 81 | (5) |
| 6 | Ph(CH2)2 | p-MeOC6H4 | f | 71 | (0) |
| 7 | Ph(CH2)2 | p-BrC6H4 | g | 87 | (4) |
| 8 | Ph | Ph(CH2)2 | h | 55 2 | (0) |
| 9 | Ph | Et | i | 54 2 | (0) |

| Time | Isolated Yield 1,2 (%) | Recovery 1 | |||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Comp. | (h) | 4 | 5 | (%) |
| 1 3 | Ph | Ph | a | 96 | 73 | (16) [88:12] | (0) |
| 2 | Ph | Ph | a | 6 | 69 | 27 [74:26] | (0) |
| 3 5 | Ph | Ph | a | 2 | (15) | (59) | (20) |
| 4 5 | Ph | Ph | a | 3 | (0) | 91 [68:32] | (0) |
| 5 3 | p-MeOC6H4 | Ph | b | 24 | (10) | (0) | (88) |
| 6 4 | p-MeOC6H4 | Ph | b | 48 | 52 | (7) | (31) |
| 7 | p-MeOC6H4 | Ph | b | 24 | (15) | 85 [73:27] | (0) |
| 8 5 | p-MeOC6H4 | Ph | b | 15 | (0) | 88 [66:34] | (0) |
| 9 3 | p-FC6H4 | Ph | c | 48 | 62 | (0) | (31) |
| 10 3 | p-FC6H4 | Ph | c | 96 | 84 | (6) | (0) |
| 11 5 | p-FC6H4 | Ph | c | 3 | (0) | 91 [65:35] | (0) |
| 12 3 | Et | Ph | d | 24 | (3) | (0) | (96) |
| 13 | Et | Ph | d | 48 | 59 | (17) | (16) |
| 14 5 | Et | Ph | d | 48 | (0) | 87 [55:45] | (trace) |
| 14 3 | Ph(CH2)2 | Ph | e | 24 | (5) | (0) | (88) |
| 15 | Ph(CH2)2 | Ph | e | 24 | 62 | (5) | (19) |
| 16 5 | Ph(CH2)2 | Ph | e | 18 | (5) | 84 [55:45] | (2) |
| 17 3 | Ph(CH2)2 | p-MeOC6H4 | f | 24 | (0) | (0) | (quant) |
| 18 | Ph(CH2)2 | p-MeOC6H4 | f | 48 | 42 | (13) | (43) |
| 19 5 | Ph(CH2)2 | p-MeOC6H4 | f | 48 | (0) | 56 [50:50] | (34) |
| 20 3 | Ph(CH2)2 | p-BrC6H4 | g | 24 | (30) | (0) | (69) |
| 21 3 | Ph(CH2)2 | p-BrC6H4 | g | 96 | 99 | (0) | (0) |
| 22 | Ph(CH2)2 | p-BrC6H4 | g | 24 | 79 | (13) | (0) |
| 23 5 | Ph(CH2)2 | p-BrC6H4 | g | 10 | (0) | 85 [55:45] | (0) |

| Temp. | Time | Isolated Yield 1,2 (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R1 | Comp. | Solvent | Additive | (°C) | (h) | 5 | 2 |
| 1 | Ph | a | DCM | TiCl4 (20) | 0 | 6 | (24) [67:33] | (71) |
| 2 | Ph | a | DCM | BF3·OEt2 (20) | 0 | 6 | (2) [–] | (68) |
| 3 3 | Ph | a | DCM | AlCl3 (20) | 0 | 6 | (6) [83:17] | (45) |
| 4 3 | Ph | a | DCM | TiCl4 (20) | −80 | 6 | (0) | (99) |
| 5 | Ph | a | DCM | Sc(OTf)3 (20) | 0 | 6 | (1) [–] | (40) |
| 6 | Ph | a | HFIP | 25 | 72 | (66) [76:24] | (6) | |
| 7 | Ph | a | THF | [PdCl2·(PhCN)2] (10) | 25 | 5 | (77) [92:8] | (2) |
| 8 | Ph | a | CH2Cl2 | [PdCl2·(PhCN)2] (10) | 25 | 5 | (80) [90:10] | (12) |
| 9 3 | Ph | a | Toluene | [PdCl2·(PhCN)2] (10) | 25 | 5 | 70 [95:5] | (0) |
| 10 3 | p-MeOC6H4 | b | Toluene | [PdCl2·(PhCN)2] (10) | 25 | 5 | 81 [95:5] | (2) |
| 11 3 | p-FC6H4 | c | Toluene | [PdCl2·(PhCN)2] (10) | 25 | 5 | 65 [94:6] | (0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamada, Y.; Matsunaga, R.; Kawasaki-Takasuka, T.; Yamazaki, T. Base-Mediated Claisen Rearrangement of CF3-Containing Bisallyl Ethers. Molecules 2021, 26, 4365. https://doi.org/10.3390/molecules26144365
Hamada Y, Matsunaga R, Kawasaki-Takasuka T, Yamazaki T. Base-Mediated Claisen Rearrangement of CF3-Containing Bisallyl Ethers. Molecules. 2021; 26(14):4365. https://doi.org/10.3390/molecules26144365
Chicago/Turabian StyleHamada, Yoko, Rio Matsunaga, Tomoko Kawasaki-Takasuka, and Takashi Yamazaki. 2021. "Base-Mediated Claisen Rearrangement of CF3-Containing Bisallyl Ethers" Molecules 26, no. 14: 4365. https://doi.org/10.3390/molecules26144365
APA StyleHamada, Y., Matsunaga, R., Kawasaki-Takasuka, T., & Yamazaki, T. (2021). Base-Mediated Claisen Rearrangement of CF3-Containing Bisallyl Ethers. Molecules, 26(14), 4365. https://doi.org/10.3390/molecules26144365