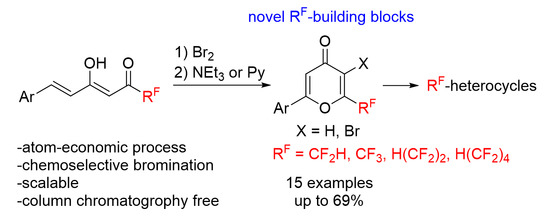
2. Results and Discussion
We started our research with the preparation of fluorinated enediones
2 based on the condensation reaction of 4-aryl-3-buten-2-ones
1 with ethyl polyfluoroalkanoates in the presence of sodium hydride in diethyl ether at room temperature or lithium hydride in benzene under reflux (
Scheme 2,
Table 1). Both methods were comparably effective, and CF
3-bearing products
2a–
i were obtained in 51–92% yields. Compounds
2a,
f–
h have been described in the literature, but they were prepared by other methods [
38,
39,
40,
41]. Besides, this approach was applied for the preparation of polyfluorinated enediones
2j–l in 43–51%.
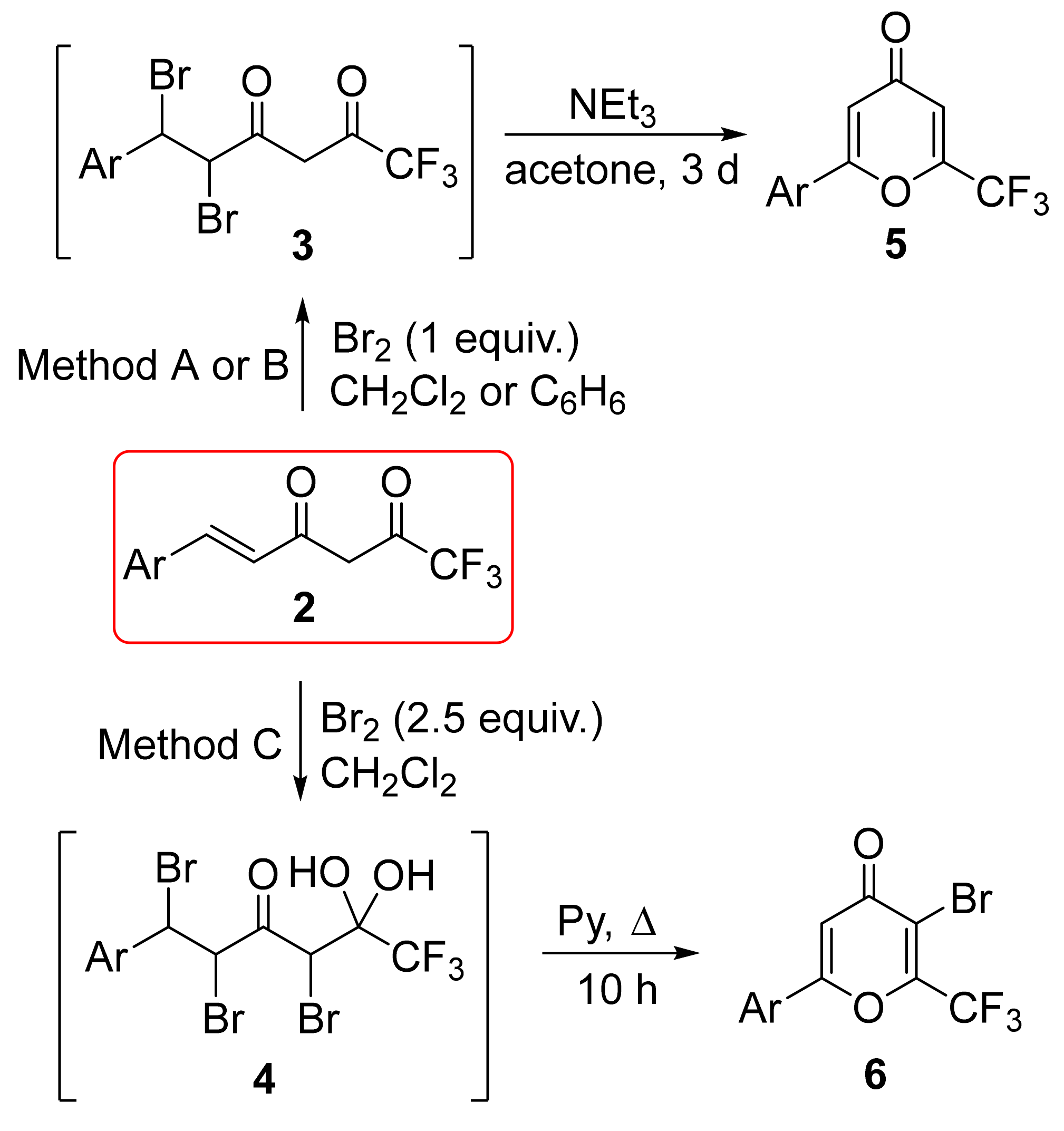
We first focused on the bromination of CF
3-containing compounds
2a–i in order to obtain active intermediates and to develop a general method for the construction of pyrones based on enediones
2. Both the double bond and the diketone moiety in 6-aryl-1,1,1-trifluorohex-5-ene-2,4-diones
2 can undergo an electrophilic attack of bromine. To estimate the influence of conditions on conversion, the chemo- and stereoselectivity (see detailed data in
Supplementary Materials), the bromination reaction was examined for compound
2b bearing the
para-fluorophenyl substituent (
Scheme 3,
Table 2). It was shown that the most active fragment is the double bond that leads to the initial formation of dibromo derivative
3b, and no product of initial attack only at the methylene group of the diketone moiety was found. At the same time, compounds
3 due to the absence of conjugation were able to react with Br
2 by the diketone moiety at a comparable rate with the starting enediones
2 giving tribromo derivatives
4.
When 1 equiv. of bromine was used, incomplete conversion was usually observed, accompanied by the formation of tribromo derivative 4b. Bromination in acetic acid gave low conversion (26%) and predominant formation of the tribromo derivative (3b:4b = 9:17) (entry 1). Carrying out the reaction in aprotic polar solvents CH2Cl2 and t-BuOMe made it possible to increase the conversion to 58 and 83%, respectively, but the selectivity turned out to be low (entries 2,4). The use of non-polar aprotic solvents (dioxane, CS2) led to the improved 3b:4b ratio and 71–75% conversion (entries 5,6). When the reaction was carried out in benzene, the highest conversion (88%), the highest NMR yield of 3b, and the good selectivity of the reaction were found (3b:4b = 78:10) (entry 7). A slight increase in the amount of bromine (up to 1.2 equiv.) led to a decrease in selectivity (3b:4b = 79:21), although the starting compound 2b was not detected at all (entry 8).
Further investigation showed that the bromination reaction is very sensitive to the nature of the substituents in the aromatic ring (
Scheme 3,
Table 2, and
Table S1 in Supplementary Materials). Thus, the reaction proceeded with the highest selectivity with one equivalent of bromine in benzene for compounds bearing moderate and good
π-donors (4-MeO, 4-F or Ar = 2-Th) (entries 7,10,11). The introduction of electron-withdrawing substituents (4-Cl, 3-NO
2) into the aromatic ring decreased the selectivity of the transformation for compounds
2c,e due to incomplete conversion and the formation of tribromo derivatives
4 as major products (entries 12,13). Even in the case of enedione
2a (Ar = Ph), selectivity and conversion of bromination in benzene turned out to be poor (entry 14), however, the use of CH
2Cl
2 as a solvent improved the selectivity and the conversion (entry 15). The introduction of the strong electron-donating substituent (4-Me
2N) led to preferential bromination at the aromatic ring rather than at the double bond followed by cyclization because of basic properties of the substituent (
Scheme S1 in Supplementary Materials). It should be noted that, in all cases, compounds
3 were formed as a sole
anti-dibromo diastereoisomer according to the classical concept of electrophilic addition of bromine to alkenes via the cyclic bromonium cation [
42].
Further, we decided to develop a procedure for the bromination of both fragments of enediones
2 simultaneously for the selective preparation of tribromo derivatives
4. The reaction of enedione
2b with an excess of bromine (2.2 equiv.) in benzene gave products with low selectivity (
3b:
4b = 56:44) (
Table 2, entry 9). Moreover, significant amounts of corresponding dihydropyrones were formed as a result of spontaneous acid-catalyzed cyclization. One of the highest yields of compound
4b in reaction with 1 equiv. of bromine was observed in dichloromethane (entry 2). The use of bromine (2.5 equiv.) in this solvent led to the almost selective formation of tribromo derivative
4b as a mixture of two diastereoisomers in a ratio of 80:20 (entry 3).
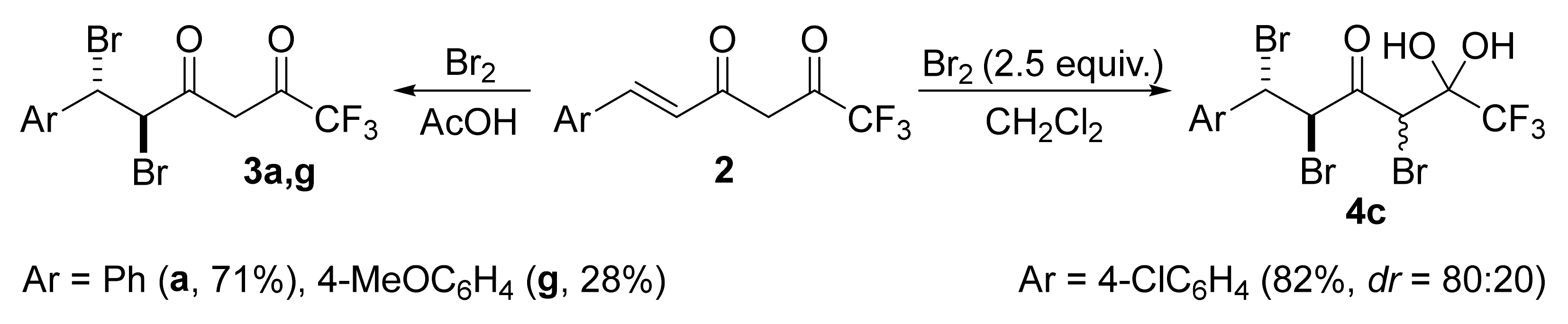
Although the reaction of enediones
2 with Br
2 gave dibromo derivatives
3 or tribromo derivatives
4 as major products, we did not usually isolate them in pure form to avoid additional losses because of their ability to cyclize and high reactivity. But for several cases, these compounds were obtained as a single isomer by recrystallization. The reaction of enediones
2a,g with one equivalent of bromine in acetic acid made it possible to obtain dibromo derivatives
3a and
3g in 71 and 28% yields, respectively (
Scheme 4).
The reaction of chloro-substituted enedione 2c with an excess of bromine (2.5 equiv.) in CH2Cl2 led to tribromo derivative 4c (dr = 80:20) smoothly in 82% yield. Compound 4c in pure crystalline form prepared by recrystallization from hexane is stable and can be stored for a long time without changes.
The structure of tribromo derivatives
4 was established on the basis of elemental analysis data and
1H and
19F NMR spectra (
Table S3 in Supplementary Materials). In the
1H NMR spectra of these compounds in CDCl
3, two singlets of the diastereotopic OH groups were observed at
δ 4.06–4.46 and 4.62–4.79 ppm. The
19F NMR spectra exhibit the CF
3 group at
δ 79.7–79.9 ppm indicating its location at the saturated carbon atom. For compounds
2 and
3, the characteristic chemical shifts of the trifluoromethyl group appeared at
δ 84.5–85.0 and 86.0 ppm, respectively (
Table S2 in Supplementary Materials).
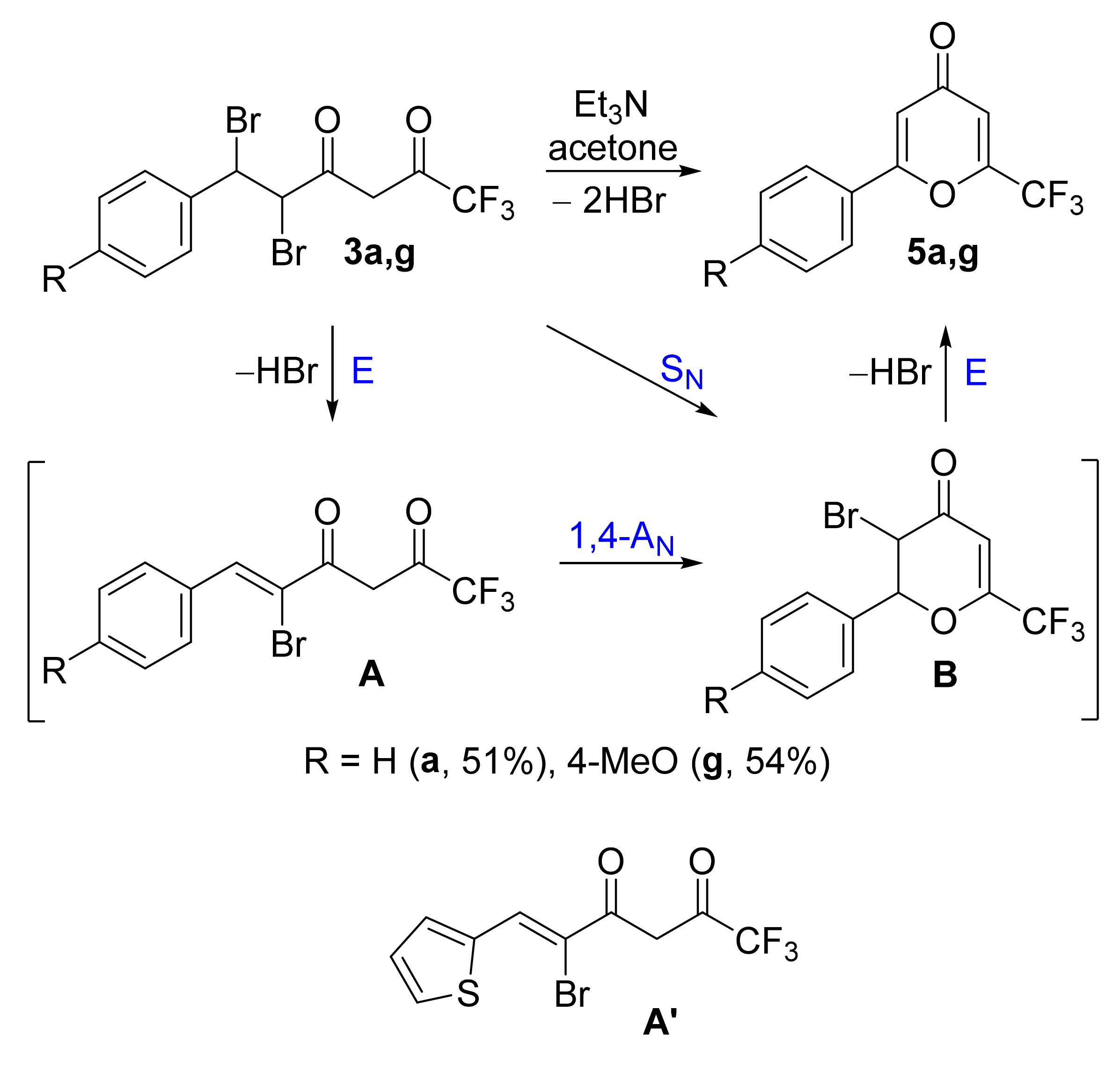
At the next stage of our work, we studied the cyclization of dibromo derivatives
3a,g in the presence of bases. The use of
t-BuOK in THF or pyridine, DIPEA, DBU, NaHCO
3 in various solvents (acetone, ethanol, DMSO) at room temperature did not allow us to obtain 4-pyrone
5a in an acceptable yield. The most favorable conditions turned out to be stirring with a 2.5-fold excess of triethylamine in acetone for three days (
Scheme 5). In addition, the isolation of the product, in this case, does not require chromatography, and pyrones
5a,g precipitated in pure form upon dilution with water in 51–54% yields. This approach can be scaled up to 5 g of enedione
2a, and the yield of pyrone
5a was 36% in two stages.
The cyclization mechanism of compounds
3 includes the formation of dihydropyran intermediate
B via two possible pathways. The first path comprises the elimination of hydrogen bromide (E) and the production of bromoenone
A, which then undergoes an intramolecular Michael reaction (1,4-A
N). The second possibility is the direct intramolecular nucleophilic substitution of bromine (S
N). As mentioned above, the cyclic pyranic form
B was observed in the bromination reaction mixtures (
Table 2), whereas bromoenones
A were detected by NMR spectroscopy on the treatment of the dibromo derivatives
3 with pyridine at room temperature. The easy formation of 2-bromoenones from 2,3-dibromoketones is also well presented in the literature [
42]. Thus, it can be assumed that the first path is more preferable in basic conditions. Intermediate
A′ was isolated in pure form with the use of column chromatography (56% yield) from enedione
2i via bromination (1 equiv.) and subsequent monodehydrobromination by DIPEA at room temperature.
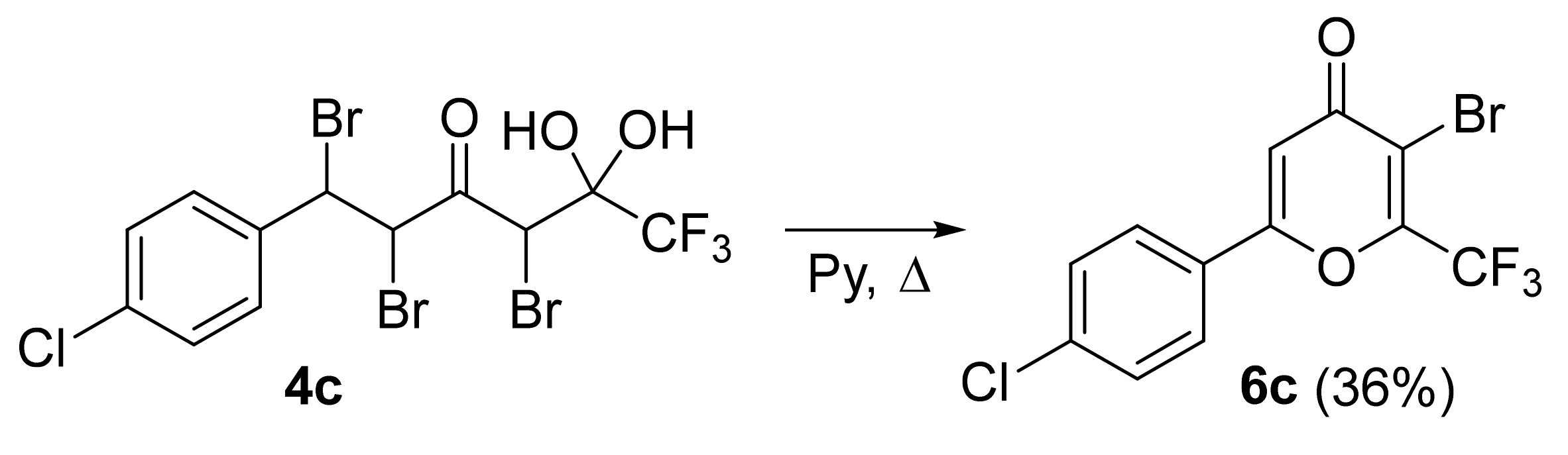
Tribromo derivatives
4 undergo cyclization under the action of triethylamine in acetone at room temperature much slower, furthermore, the high basicity of the medium could provide a possibility of side reactions associated with detrifluoroacetylation [
41]. Therefore, dehydrohalogenation of
4 was carried out by refluxing for 10 h in pyridine as a solvent and a weaker base (
Scheme 6). The cyclization of compound
4c gave pyrone
6c in 36% yield. These results demonstrate that the cyclization step determines mainly total output of the pyrones. Moreover, the difference in reaction rates of cyclization of tribromo derivatives
4 and dibromo derivatives
3 allowed the isolation of pyrones
5 in the presence of Et
3N in acetone selectively, even when the reaction mixtures of the brominated intermediates were used.
With the optimized conditions of the bromination and cyclization of intermediate bromo derivatives in hand, we decided to develop a simple
one-pot two-step preparative method for the synthesis of 4-pyrones
5 and
6 based on oxidative cyclization of enediones
2. Considering that the reaction mixtures of bromination contain various bromine-bearing forms (cyclic and acyclic), the overall efficiency and convenience of this two-step process without intermediate purification can improve (
Scheme 7,
Table 3).
For the preparation of 4-pyrones 5 in the one-pot approach, enediones 2 were treated with 1 equiv. of bromine in benzene (Method A) for compounds bearing electron-donating substituents (F, Me, MeO) and in CH2Cl2 (Method B) for compounds bearing weak electron-withdrawing substituents (Cl, Br) or H at the C-4 position. After bromination, the solvent was evaporated, and the cyclization was carried out using Et3N (2.5 equiv.) in acetone, as a result, pyrones 5 were obtained in 28–69% yields.
The preparation of 3-bromo-4-pyrones 6 with a one-pot method was possible only for substrates bearing weak electron-donating (H, 4-Me, 4-F) and electron-withdrawing groups (4-Cl, 3-NO2) in the benzene ring since aromatic bromination was unavoidable with an excess of bromine in case of highly activated aryl substituents. At the first stage, enedione 2 was treated with 2.5 equiv. of bromine in CH2Cl2 and subsequent heating at 100 °C in pyridine for 10 h led to the formation of pyrones 6 in 4–42% yields (Method C).
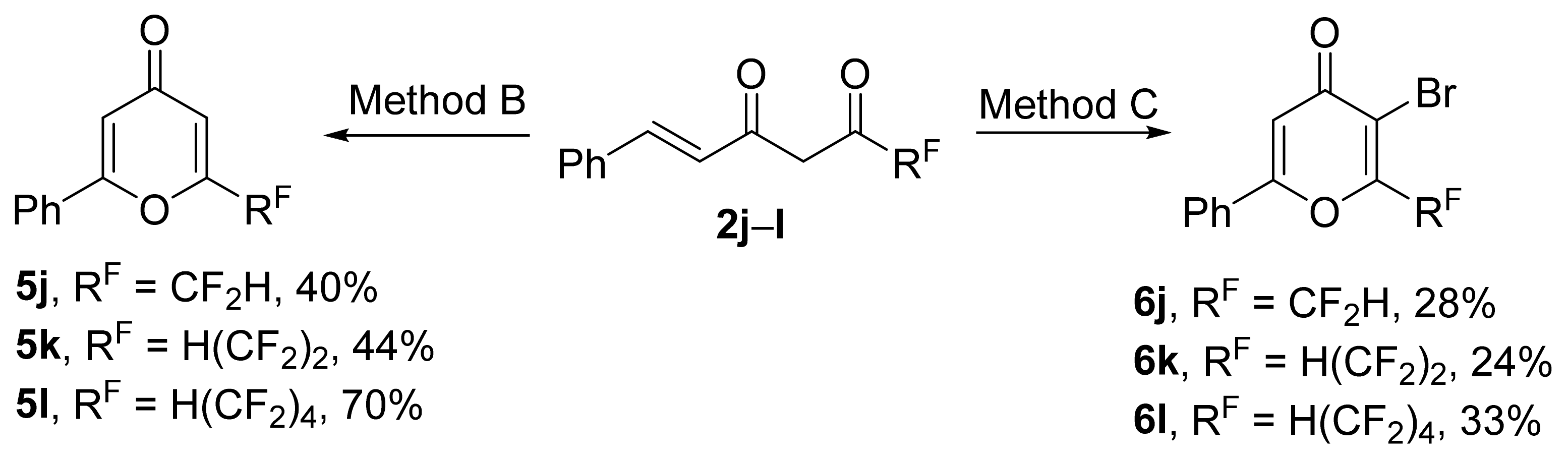
This
one-pot approach was also applied for the preparation of phenyl-4-pyrones bearing various polyfluoroalkyl substituent. No significant difference in reactivity was observed depending on fluorine content or chain length of R
F-moiety, and the cyclization of enediones
2j–
l in the presence of 1 equiv. of bromine (Method B) gave 4-pyrones
5j–l in 40–70% yields (
Scheme 8). The use of Method C led to 3-bromopyrones
6j–l in 24–33% yields.
The structure of 4-pyrones 5 and 6 was established on the basis of elemental analysis data, HRMS and NMR spectra. In the 1H NMR spectra of compounds 5 in CDCl3, characteristic doublets of H-3 and H-5 protons of the pyrone ring were observed at δ 6.63–6.88 ppm with a coupling constant of 1.8–2.2 Hz. In the case of pyrones 6, the signal of the H-5 proton of the pyrone ring appeared as a singlet at δ 6.88–7.04 ppm (CDCl3).
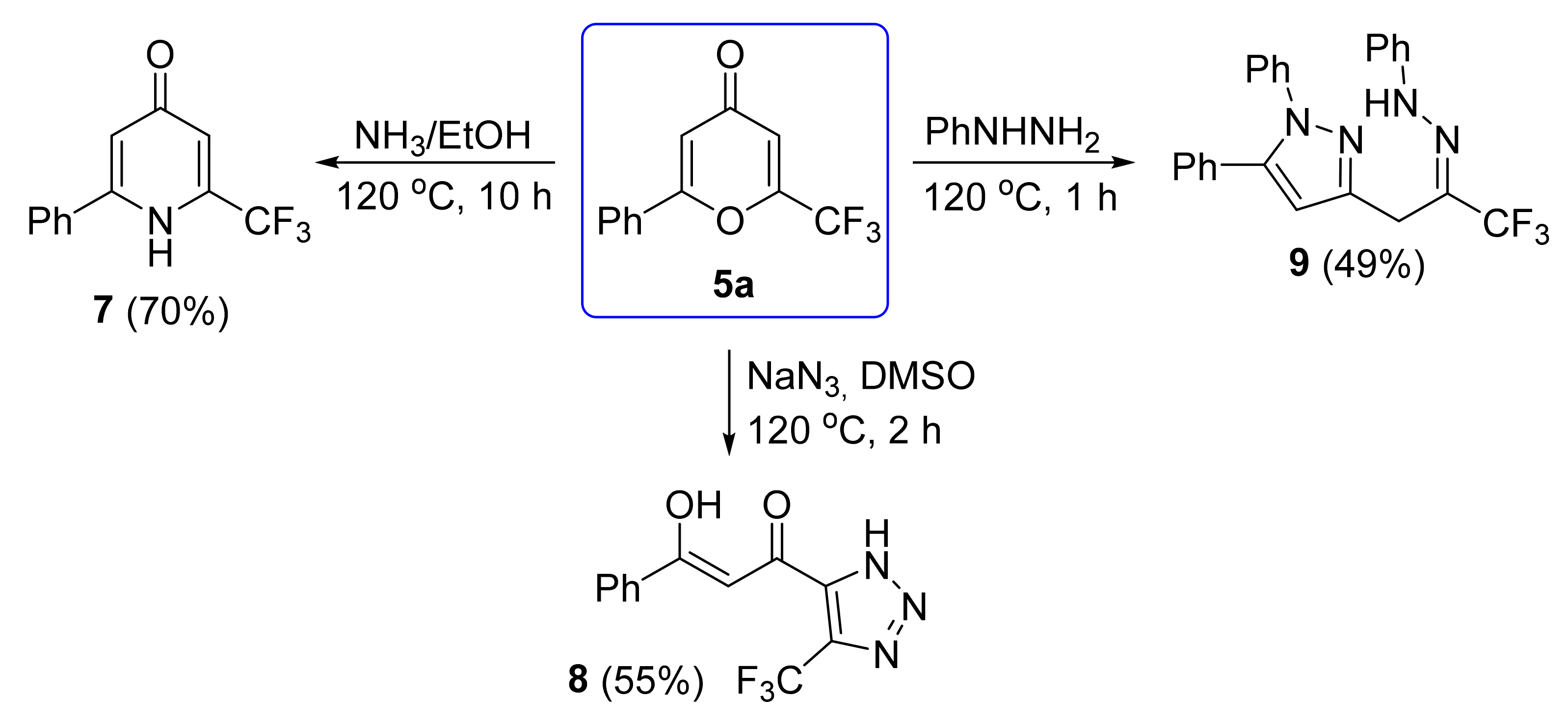
The next step was to study the chemical properties of 2-aryl-6-(trifluoromethyl)-4-pyrones with N-nucleophiles to obtain azaheterocycles (
Scheme 9). The reaction of pyrone
5a with ammonia in ethanol in an autoclave at 120 °C for 10 h led to the formation of pyridone
7 in 70% yield. The ring-opening transformation of pyrone
5a with sodium azide [
43] in DMSO afforded triazole
8 in 55% yield. Phenylhydrazine reacted with pyrone
5a regioselectively attacking with the amino group at the C-2 and C-4 positions to give pyrazole
9. The structure of pyrazole
9 was assigned based on NMR spectra according to the literature data [
16,
34].
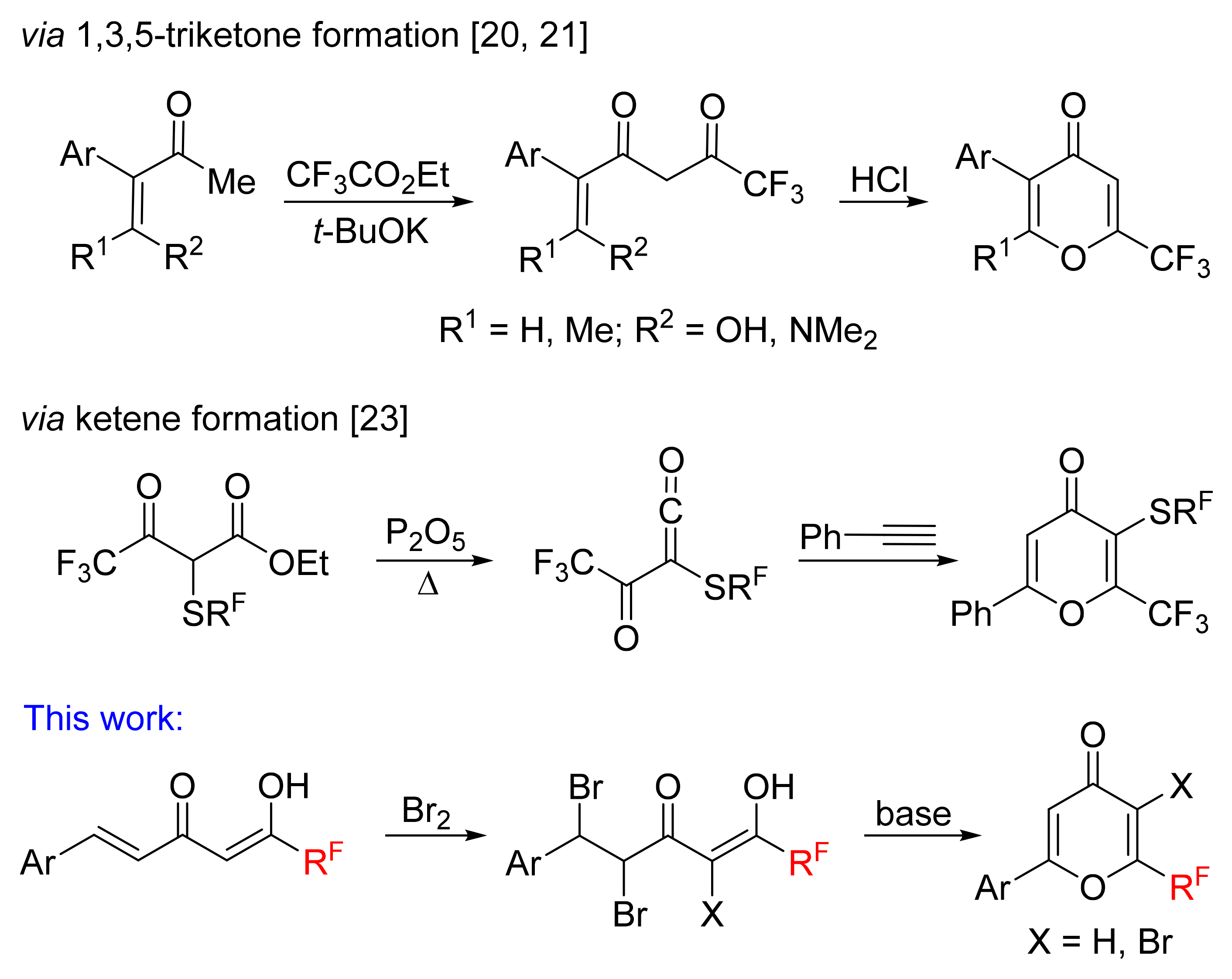
Thus, a convenient and general method for the synthesis of 2-aryl-6-polyfluoroalkyl-4-pyrones and their bromo derivatives has been developed based on one-pot oxidative cyclization of fluorinated enediones. The selectivity of monobromination of 6-aryl-1,1,1-trifluorohex-5-ene-2,4-diones is connected with the predominant electrophilic attack on the double bond and strongly depends on the solvent used and the nature of the substituent in the aromatic ring, which determines the scope of obtained fluorinated 2-aryl-4-pyrones. 6-Aryl-1,1,1-trifluorohex-5-ene-2,4-diones bearing electron-withdrawing and weak electron-donating substituents in the aromatic ring react with an excess of bromine leading to tribromo derivatives as a result of bromination of the double bond and the diketone moiety. Di- and tribromo derivatives in a basic medium selectively undergo cyclization to form 4-pyrones. The resulting 4-pyrones have proven to be useful building blocks and have been utilized to synthesize CF3-bearing heterocycles, which are of further interest in terms of potential biological activity.
3. Materials and Methods
NMR spectra were recorded on Bruker DRX-400 (
1H: 400 MHz,
13C: 100 MHz,
19F: 376.5 MHz) and Bruker Avance III-500 (
1H: 500 MHz,
13C: 126 MHz,
19F: 471 MHz) spectrometers in DMSO-
d6 and CDCl
3. The chemical shifts (
δ) are reported in ppm relative to the internal standard TMS (
1H NMR), C
6F
6 (
19F NMR), and residual signals of the solvents (
13C NMR). IR spectra were recorded on a Shimadzu IRSpirit-T spectrometer using an attenuated total reflectance (ATR) unit (FTIR mode, ZnSe crystal), the absorbance maxima (
ν) are reported in cm
–1. Elemental analyses were performed on an automatic analyzer PerkinElmer PE 2400. Melting points were determined using a Stuart SMP40 melting point apparatus. Column chromatography was performed on silica gel (Merck 60, 70–230 mesh). All solvents that were used were dried and distilled by standard procedures. Arylideneacetones
1 have been synthesized according to the procedure, previously described in the literature [
44].
3.1. Synthesis of Enediones 2a–l
General Procedure. A suspension of NaH (60% by weight in mineral oil) (240 mg, 6.0 mmol) in dry Et2O (5 mL) was cooled on an ice bath, then CF3CO2Et (0.86 g, 6.0 mmol) and a solution of arylideneacetone 1 (4.0 mmol) in dry Et2O (3 mL) were successively added under stirring. The reaction mixture was warmed to room temperature and stirred for 20 h. Then 1M aqueous HCl (10 mL) was added, and the organic phase was separated (if the precipitate of diketonate formed, it was filtered and treated with 1M aqueous HCl (10 mL)). The aqueous phase was extracted with AcOEt (5 mL). The combined organics were washed with H2O and dried over anhydrous Na2SO4, the solvents evaporated, and the residue recrystallized from hexane with decantation of hot solution from insoluble tarry by-products.
General procedure (with LiH) [
38]. A solution of arylideneacetone 1 (4.0 mmol) in dry C
6H
6 (1 mL) and CF
3CO
2Et (0.86 g, 6.0 mmol) were successively added to a suspension of LiH (48 mg, 6.0 mmol) in dry C
6H
6 (4 mL) under stirring. The reaction mixture was warmed until the reaction started and then was stirred at room temperature for 20 h. After that, the reaction mixture was poured into a mixture of ice (4 g) and H
2SO
4 (1 mL), extracted with chloroform (5 mL), and dried over MgSO
4. The solvents were evaporated, and the residue recrystallized from hexane with decantation of hot solution from insoluble tarry by-products.
(E)-1,1,1-Trifluoro-6-phenylhex-5-ene-2,4-dione (
2a). Yield 0.73 g (75%), colorless needles, mp 56–58 °C (lit. mp 58 °C [
40], 61–61.5 °C [
38]).
1H NMR (400 MHz, CDCl
3) enol form:
δ 6.04 (1H, s, 3-CH); 6.58 (1H, d,
J = 15.9 Hz, 5-CH); 7.37–7.57 (3H, m, H Ph); 7.53–7.61 (2H, m, H-2, H-6 Ph); 7.77 (1H, d,
J = 15.9 Hz, 6-CH); 14.22 (1H, s, OH). The analytical data are in consistence with the literature [
38,
40].
(E)-1,1,1-Trifluoro-6-(4-fluorophenyl)hex-5-ene-2,4-dione (2b). Yield 0.67 g (64%), yellow prisms, mp 83–84 °C. IR (ATR) ν 1637, 1578, 1509, 1444, 1414. 1H NMR (500 MHz, CDCl3) enol form: δ 6.02 (1H, s, 3-CH); 6.50 (1H, d, J = 15.8 Hz, 5-CH); 7.12 (2H, t, J = 8.6 Hz, JHF = 8.6 Hz, H-3, H-5 Ar); 7.57 (2H, dd, J = 8.7 Hz, J = 5.3 Hz, H-2, H-6 Ar); 7.73 (1H, d, J = 15.8 Hz, 6-CH); 14.24 (1H, s, OH). 19F NMR (376 MHz, CDCl3) δ 53.9 (tt, JHF = 8.4 Hz, JHF = 5.4 Hz, F); 84.7 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 95.6 (q, 3JCF = 1.4 Hz, C-3); 116.4 (d, 2JCF = 22.0 Hz, C-3, C-5 Ar); 116.7 (q, 1JCF = 286.6 Hz, CF3); 120.8 (d, 6JCF = 2.4 Hz, C-5); 130.4 (s, C-1 Ar); 130.5 (d, 4JCF = 8.8 Hz, C-2, C-6 Ar); 142.3 (C-6); 164.3 (d, 1JCF = 253.2 Hz, C-4 Ar); 180.5 (q, 2JCF = 36.1 Hz, C-2); 180.8 (C-4). HRMS (ESI) m/z [M + H]+. Calcd for C12H9F4O2: 261.0539. Found: 261.0533.
(E)-6-(4-Chlorophenyl)-1,1,1-trifluorohex-5-ene-2,4-dione (2c). Yield 0.66 g (60%), yellow needles, mp 92–93 °C. IR (ATR) ν 3072, 3035, 1632, 1610, 1573, 1561, 1491, 1454, 1410. 1H NMR (500 MHz, CDCl3) enol form: δ 6.03 (1H, s, 3-CH); 6.54 (1H, d, J = 15.8 Hz, 5-CH); 7.40 (2H, d, J = 8.5 Hz, H Ar); 7.50 (2H, d, J = 8.5 Hz, H Ar); 7.71 (1H, d, J = 15.8 Hz, 6-CH); 14.17 (1H, s, OH). 19F NMR (471 MHz, CDCl3) δ 84.6 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 95.80 (q, 3JCF = 1.4 Hz, C-3); 116.63 (q, 1JCF = 285.6 Hz, CF3); 121.5 (s, C-5); 129.4 (s, 2H); 129.6 (s, 2H); 132.7 (s, C Ar); 137.1 (s, C Ar); 142.1 (s, C-6); 180.4 (s, C-4); 180.8 (q, 2JCF = 36.1 Hz, C-2). Anal. Calcd for C12H8ClF3O2: C 52.10; H 2.91. Found: C 51.93; H 2.94%.
(E)-6-(4-Bromophenyl)-1,1,1-trifluorohex-5-ene-2,4-dione (2d). Yield 1.18 g (92%), white crystals, mp 86–87 °C. IR (ATR) ν 1632, 1609, 1582, 1487, 1454, 1406. 1H NMR (400 MHz, CDCl3) enol form: δ 6.03 (1H, s, 3-CH); 6.56 (1H, d, 3J = 15.9 Hz, 5-CH); 7.43 (2H, d, 3J = 8.4 Hz, H Ar); 7.56 (2H, d, 3J = 8.4 Hz, H Ar); 7.69 (1H, d, 3J = 15.9 Hz, 6-CH); 14.15 (1H, s, OH). 19F NMR (376 MHz, CDCl3) δ 84.7 (s, CF3). 13C NMR (101 MHz, CDCl3) δ 95.8 (unresolved q, C-3); 116.7 (q, 1JCF = 285.5 Hz, CF3); 121.6 (C-5); 125.5 (C Ar); 129.8 (2C Ar); 132.4 (2C Ar); 133.1 (C Ar); 142.1 (C-6); 180.4 (C-4); 180.8 (q, 2JCF = 36.2 Hz, C-2). Anal. Calcd for C12H8BrF3O2: C 44.89; H 2.51. Found: C 45.03; H 2.57%.
(E)-1,1,1-Trifluoro-6-(3-nitrophenyl)hex-5-ene-2,4-dione (2e). Yield 0.59 g (51%), white scales, mp 118–119 °C. IR (ATR) ν 1644, 1610, 1577, 1523, 1477, 1429. 1H NMR (400 MHz, CDCl3) enol form: δ 6.10 (1H, s, 3-CH); 6.70 (1H, d, 3J = 15.9 Hz, 5-CH); 7.63 (1H, t, 3J = 8.0 Hz, H-5 Ar); 7.79 (1H, d, 3J = 15.9 Hz, 6-CH); 7.86 (1H, dt, 3J = 7.7 Hz, 4J = 1.0 Hz, H-6 Ar); 8.27 (1H, ddd, 3J = 8.5 Hz, 4J = 2.1 Hz, 4J = 0.8 Hz, H-4 Ar); 8.44 (1H, t, 4J = 1.8 Hz, H-2 Ar); 13.96 (1H, s, OH). 19F NMR (376 MHz, CDCl3) δ 84.5 (s, CF3). HRMS (ESI) m/z [M – H]–. Calcd for C12H7F3NO4: 286.0327. Found: 286.0331.
(E)-1,1,1-Trifluoro-6-(p-tolyl)hex-5-ene-2,4-dione (
2f). Yield 0.67 g (65%), yellow needles, mp 61–62 °C. IR (ATR) ν 3031, 2956, 2926, 2360, 1652, 1594, 1575, 1516, 1447, 1416.
1H NMR (400 MHz, CDCl
3) enol form:
δ 2.40 (3H, s, Me); 6.01 (1H, s, 3-CH); 6.53 (1H, d,
J = 15.8 Hz, 5-CH); 7.23 (2H, d,
J = 8.0 Hz, H-3, H-5 Ar); 7.47 (2H, d,
J = 8.1 Hz, H-2, H-6 Ar); 7.75 (1H, d,
J = 15.8 Hz, 6-CH); 14.30 (1H, s, OH).
19F NMR (376 MHz, CDCl
3)
δ 84.7 (s, CF
3).
13C NMR (101 MHz, CDCl
3)
δ 21.6 (CH
3); 95.4 (q,
3JCF = 1.5 Hz, C-3); 116.8 (q,
1JCF = 285.3 Hz, CF
3); 120.0 (C-5); 128.6 (2C Ar); 129.9 (2C Ar); 131.5 (C Ar); 141.9 (C Ar); 143.90 (C-6); 180.1 (q,
2JCF = 36.0 Hz, C-2); 181.5 (C-4). Anal. Calcd for C
13H
11F
3O
2: C 60.94; H 4.33. Found: C 60.96; H 4.47%. The analytical data are in consistence with the literature [
41].
(E)-1,1,1-Trifluoro-6-(4-methoxyphenyl)hex-5-ene-2,4-dione (
2g). Yield 0.88 g (81%), yellow needles, mp 100–101 °C (lit. mp 98 °C [
40]). IR (ATR) ν 1633, 1568, 1511, 1453, 1441, 1420.
1H NMR (500 MHz, CDCl
3) enol form:
δ 3.86 (3H, s, OMe); 6.00 (1H, s, 3-CH); 6.45 (1H, d,
3J = 15.8 Hz, 5-CH); 6.94 (2H, d,
3J = 8.8 Hz, H-3, H-5 Ar); 7.53 (2H, d,
3J = 8.4 Hz, H-2, H-6 Ar); 7.74 (1H, d,
3J = 15.8 Hz, 6-CH); 14.45 (1H, s, OH).
19F NMR (471 MHz, CDCl
3)
δ 84.8 (s, CF
3).
13C NMR (101 MHz, CDCl
3)
δ 55.5 (OMe); 95.2 (q,
2JCF = 1.2 Hz, C-3); 114.7 (2C Ar); 116.9 (q,
1JCF = 285.1 Hz, CF
3); 118.6 (C-5); 126.9 (C Ar); 130.5 (2C Ar); 143.7 (C-6); 162.2 (C Ar); 179.7 (q,
2JCF = 35.9 Hz, C-2); 181.9 (C-4). Anal. Calcd for C
13H
11F
3O
3: C 57.36; H 4.07. Found: C 57.38; H 4.20%. The analytical data are in consistence with the literature [
40].
(E)-6-(4-(Dimethylamino)phenyl)-1,1,1-trifluorohex-5-ene-2,4-dione (
2h). Yield 0.76 g (67%), magenta needles, mp 79–80 °C (lit. mp 78–79 °C [
39]). IR (ATR) ν 2900, 1556, 1519, 1453, 1430, 1360, 1328.
1H NMR (500 MHz, CDCl
3) enol form:
δ 3.07 (6H, s, NMe
2); 5.95 (1H, s, 3-CH); 6.35 (1H, d,
3J = 15.6 Hz, 5-CH); 6.68 (2H, d,
3J = 8.9 Hz, H-3, H-5 Ar); 7.47 (2H, d,
3J = 8.9 Hz, H-2, H-6 Ar); 7.74 (1H, d,
3J = 15.6 Hz, 6-CH); 14.69 (1H, s, OH).
19F NMR (471 MHz, CDCl
3)
δ 85.0 (s, CF
3). Anal. Calcd for C
14H
14F
3NO
2: C 58.95; H 4.95; N 4.91. Found: C 58.92; H 4.94; N 4.79%. The analytical data are in consistence with the literature [
39].
(E)-1,1,1-Trifluoro-6-(thiophen-2-yl)hex-5-ene-2,4-dione (2i). Yield 0.83 g (84%), yellow rectangular prisms, mp 94–95 °C. IR (ATR) ν 1577, 1503, 1437, 1416. 1H NMR (400 MHz, CDCl3) enol form: δ 5.99 (1H, s, 3-CH); 6.36 (1H, d, 3J = 15.5 Hz, 5-CH); 7.11 (1H, dd, 3J = 4.9 Hz, 3J = 3.8 Hz, H-4 Th); 7.35 (1H, d, 3J = 3.5 Hz, H-3 Th); 7.48 (1H, d, 3J = 5.0 Hz, H-5 Th); 7.88 (1H, d, 3J = 15.5 Hz, 6-CH); 14.28 (1H, s, OH). 19F NMR (376 MHz, CDCl3) δ 84.8 (s, CF3). 13C NMR (101 MHz, CDCl3) δ 95.5 (unresolved q, C-3); 116.8 (q, 1JCF = 285.3 Hz, CF3); 119.8 (C-5); 128.6; 130.2; 132.4; 136.2; 139.8; 180.1 (q, 2JCF = 36.0 Hz, C-2); 180.8 (C-4). Anal. Calcd for C10H7F3O2S: C 48.39; H 2.84. Found: C 48.36; H 2.75%.
(E)-1,1-Difluoro-6-phenylhex-5-ene-2,4-dione (2j). Yield 0.39 g (43%), yellow crystals, mp 85–87 °C. IR (ATR) ν 3030, 1647, 1495, 1341, 1180, 1105, 867, 757. 1H NMR (500 MHz, CDCl3) enol form: δ 5.91 (1H, t, 2JHF = 54.3 Hz, CF2H); 6.04 (1H, s, 3-CH); 6.58 (1H, d, J = 15.9 Hz, 5-CH); 7.39–7.44 (3H, m, H Ph); 7.51–7.62 (2H, m, H-2, H-6 Ph); 7.72 (1H, d, J = 15.9 Hz, 6-CH); 14.58 (1H, s, OH). 19F NMR (471 MHz, CDCl3) δ 35.0 (dd, 2JHF = 54.4 Hz, 4JHF = 0.5 Hz, CF2H). 13C NMR (126 MHz, CDCl3) δ 96.0 (t, 3JCF = 1.9 Hz, C-4); 109.6 (t, 1JCF = 249.0 Hz, CF2H); 121.8 (C-6); 128.4 (2C Ph); 129.1 (2C Ph); 130.8 (C Ph); 134.4 (C Ph); 142.7 (C-7); 181.0 (C-5); 186.5 (t, 2JCF = 25.7 Hz, C-3). Anal. Calcd for C12H10F2O2: C 64.29; H 4.50. Found: C 64.69; H 4.35.
(E)-6,6,7,7-Tetrafluoro-1-phenylhept-1-ene-3,5-dione (2k). Yield 0.47 g (43%), yellow liquid. IR (ATR) ν 3028, 1633, 1580, 1449, 1244, 1110, 700. 1H NMR (400 MHz, CDCl3) enol form: δ 6.10 (tt, 2JHF = 52.9 Hz, 3JHF = 5.1 Hz, CF2H); 6.13 (1H, s, 4-CH); 6.58 (1H, d, J = 15.9 Hz, 6-CH); 7.40–7.45 (3H, m, H Ph); 7.53–7.61 (2H, m, H-2, H-6 Ph); 7.77 (1H, d, J = 15.9 Hz, 7-CH); 14.45 (1H, s, OH). 19F NMR (376 MHz, CDCl3) δ 23.4 (dt, 2JHF = 52.9 Hz, 3JFF = 6.9 Hz, CF2H); 35.8 (tdd, 3JFF = 6.9 Hz, 3JHF = 5.1 Hz, 4JHF = 0.8 Hz, CF2). 13C NMR (126 MHz, CDCl3) δ 96.8 (unresolved t, C-4); 109.1 (tt, 1JCF = 251.2 Hz, 2JCF = 34.1 Hz, CF2H); 109.5 (tt, 1JCF = 258.5 Hz, 2JCF = 27.8 Hz, CF2); 121.3 (C-6); 128.4 (2C Ph); 129.1 (2C Ph); 131.0 (C Ph); 134.3 (C Ph); 143.6 (C-7); 180.1 (q, 2JCF = 26.7 Hz, C-3); 180.9 (C-5). HRMS (ESI) m/z [M + H]+. Calcd for C13H11F4O2: 275.0695. Found: 275.0694.
(E)-6,6,7,7,8,8,9,9-Octafluoro-1-phenylnon-1-ene-3,5-dione (2l). Yield 0.76 g (51%), yellow liquid. IR (ATR) ν 2929, 1633, 1580, 1450, 1172, 1034, 972, 787. 1H NMR (500 MHz, CDCl3) δ enol form (88%): 6.08 (1H, s, 4-CH); 6.12 (1H, tt, 2JHF = 52.0 Hz, 3JHF = 5.5 Hz, CF2H); 6.58 (1H, d, J = 15.8 Hz, 2-CH); 7.39–7.47 (3H, m, H Ph); 7.54–7.61 (2H, m, H-2, H-6 Ph); 7.79 (1H, d, J = 15.8 Hz, 1-CH); 14.33 (1H, s, OH); keto form (12%): 2.77 (2H, s, CH2); 6.08 (1H, tt, 2JHF = 52.0 Hz, 3JHF = 5.5 Hz, CF2H); 6.46 (1H, d, J = 15.9 Hz, 2-CH); 7.39–7.47 (3H, m, H Ph); 7.54–7.61 (2H, m, H-2, H-6 Ph); 7.81 (1H, d, J = 15.9 Hz, 1-CH). 19F NMR (471 MHz, CDCl3) δ enol form (88%): 24.4 (dm, 2JHF = 51.9 Hz, CF2H); 32.0 –32.1 (m, 8-CF2); 37.0 (t, 3JFF = 8.2 Hz, 7-CF2); 40.5 (t, 3JFF = 9.9 Hz, 6-CF2); keto form (12%): 24.4 (dm, 2JHF = 51.9 Hz, CF2H); 31.9–32.0 (m, 8-CF2); 36.9 (t, 3JFF = 8.2 Hz, 7-CF2); 42.3 (t, 3JFF = 9.6 Hz, 6-CF2). 13C NMR (126 MHz, CDCl3) δ 97.2 (unresolved m, C-6); 107.6 (tt, 1JCF = 254.7 Hz, 2JCF = 30.9 Hz, CF2H); 107.7–113.5 (m, 3CF2); 121.0 (C-8); 128.5 (2C Ph); 129.1 (2C Ph); 131.2 (C Ph); 134.2 (C Ph); 144.0 (C-9); 180.5 (C-7); 182.5 (t, 2JCF = 26.7 Hz, C-5). HRMS (ESI) m/z [M + H]+. Calcd for C15H11F8O2: 375.0631. Found: 375.0627.
3.2. Synthesis of Compounds 3a,g
General procedure. A solution of Br2 (3.20 g, 0.020 mol) in glacial AcOH (5.7 mL) was added dropwise to enedione 2 (0.020 mol) in glacial AcOH (18 mL) cooled by a water bath. After that, the reaction mixture was stirred for 1 h at room temperature. The resulting solution was diluted by water (30 mL). The precipitate that formed was filtered. If it is needed, the pure product can be obtained by recrystallization from heptane.
(5S,6R)-5,6-Dibromo-1,1,1-trifluoro-6-phenylhexane-2,4-dione (3a). Yield 5.71 g (71%), beige powder, mp 119–120 °C. IR (ATR) ν 1665, 1629, 1604, 1497, 1458, 1434. 1H NMR (400 MHz, DMSO-d6) enol form: δ 5.56 (1H, d, 3J = 11.7 Hz, CHBr); 6.08 (1H, d, 3J = 11.7 Hz, CHBr); 6.30 (1H, s, 3-CH); 7.29–7.46 (3H, m, H Ph); 7.49–7.54 (2H, m, H-2, H-6 Ph). 1H NMR (400 MHz, CDCl3) δ 4.96 (1H, d, 3J = 11.6 Hz, CHBr); 5.39 (1H, d, 3J = 11.6 Hz, CHBr); 6.24 (1H, s, 3-CH); 7.38–7.50 (5H, m, H Ph). 19F NMR (471 MHz, CDCl3) δ 86.0 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 48.8; 51.2; 96.5; 117.1 (q, J = 280.4 Hz, CF3); 128.0; 129.0; 129.6; 137.4; 171.9 (q, J = 37.2 Hz, C-2); 190.6. HRMS (ESI) m/z [M – H]–. Calcd for C12H8Br2F3O2: 398.8843. Found: 398.8835.
(5S,6R)-5,6-Dibromo-1,1,1-trifluoro-6-(4-methoxyphenyl)hexane-2,4-dione (3g). Yield 2.42 g (28%), yellow powder, mp 124–125 °C. IR (ATR) ν 1665, 1607, 1514, 1462, 1443. 1H NMR (500 MHz, CDCl3) enol form: δ 3.84 (3H, s, OMe); 4.93 (1H, d, 3J = 11.6 Hz, CHBr); 5.37 (1H, d, 3J = 11.6 Hz, CHBr); 6.21 (1H, s, 3-CH); 6.92 (2H, d, 3J = 8.6 Hz, H-3, H-5 Ar); 7.35 (2H, d, 3J = 8.6 Hz, H-2, H-6 Ar); 13.50 (1H, s, OH). 19F NMR (376 MHz, CDCl3) δ 86.0 (s, CF3). Anal. Calcd for C13H11Br2F3O3: C 36.14; H 2.57. Found: C 36.48; H 2.73%.
3.3. Synthesis of Compound 4c
A solution of Br2 (400 mg, 2.5 mmol) in CH2Cl2 (2 mL) was added to enedione 2c (277 mg, 1 mmol) in CH2Cl2 (8 mL) under cooling on an ice-bath. The reaction mixture was left at room temperature for 15 h, and then the solvent was evaporated without heating. The residue is a mixture of diastereoisomeres (dr = 80:20), which was recrystallized from hexane to obtain single isomer in pure form for analysis.
(1Rʹ,2Sʹ)-1,2,4-Tribromo-1-(4-chlorophenyl)-6,6,6-trifluoro-5,5-dihydroxyhexan-3-one (4c). Yield 0.44 g (82%), white powder, mp 125–128 °C (toluene-hexane). After recrystallization from hexane, mp 154–155 °C (for the major diastereoisomer in pure form). IR (ATR) ν 3015, 1714, 1597, 1493, 1295, 1207, 990, 830, 715. 1H NMR (500 MHz, CDCl3) δ major diastereomer (80%): 4.38 (1H, s, OH); 4.74 (1H, s, OH); 4.95 (1H, s, 4-CHBr); 5.24 (1H, d, 3J = 11.3 Hz, 2-CHBr); 5.35 (1H, d, 3J = 11.3 Hz, 1-CHBr); 7.32–7.43 (4H, m, H Ar); minor diastereomer (20%): 3.94 (1H, s, OH); 4.58 (1H, s, OH); 4.98 (1H, s, 4-CHBr); 5.19 (1H, d, 3J = 11.3 Hz, 2-CHBr); 5.27 (1H, d, 3J = 11.3 Hz, 1-CHBr); 7.34–7.41 (4H, m, H Ar). 19F NMR (471 MHz, CDCl3) δ major diastereomer: 79.7 (s, CF3). Anal. Calcd for C12H9Br3ClF3O3: C 27.02; H 1.70. Found: C 27.32; H 1.79%.
3.4. Synthesis of Compound A’
A solution of dibromide 3i, which was prepared by reaction of enedione 2i (248 mg, 1 mmol) with Br2 (160 mg, 1 mmol) in CH2Cl2 (3.4 mL) without additional purification, and DIPEA (323 mg, 2.5 mmol) in CH2Cl2 (5 mL) was stirred at room temperature for 15 h. The reaction mixture was then quenched with aqueous HCl (10 mL, 0.5 M), the organic phase was separated, washed with water and brine, dried under Na2SO4, and evaporated. The crude product was purified by column chromatography (CH2Cl2).
5-Bromo-1,1,1-trifluoro-6-(thiophen-2-yl)hex-5-ene-2,4-dione (A’). Yield 183 mg (56%), dark yellow solid, mp 84–85 °C. IR (ATR) ν 1623, 1573, 1413. 1H NMR (500 MHz, CDCl3) enol form: δ 6.69 (1H, s, 3-CH); 7.22 (1H, dd, 3J = 4.9 Hz, 3J = 3.5 Hz, H-4 Th); 7.65 (1H, d, 3J = 3.5 Hz, H-3 Th); 7.72 (1H, d, 3J = 4.9 Hz, H-5 Th); 8.53 (1H, s, 6-CH); 14.77 (1H, s, OH). 19F NMR (471 MHz, CDCl3) δ 85.5 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 95.0 (q, 3JCF = 2.1 Hz, C-4); 112.4; 117.2 (q, 1JCF = 283.1 Hz, CF3); 127.6; 133.2; 134.1; 136.9; 137.7; 176.2 (q, 2JCF = 37.1 Hz, C-2); 182.3 (C-4). HRMS (ESI) m/z [M + H]+. Calcd for C10H7BrF3O2S: 326.9302. Found: 326.9315.
3.5. Synthesis of Pyrones 5
General procedure. A solution of Br2 (160 mg, 1.0 mmol) in the solvent (1.7 mL) was added to enedione 2 (1 mmol) in the solvent (2.7 mL) under cooling on an ice-bath (C6H6 was used for Method A, CH2Cl2 – Method B). After that, the reaction mixture was stirred at room temperature for 15 h, and then the solvent was evaporated without heating. Then acetone (4.3 mL) was added to the residue, and the solution was treated with Et3N (0.253 g, 2.5 mmol) under cooling on an ice-bath. After that, the reaction mixture was stirred at room temperature for 3 days and was diluted with H2O (15 mL). The precipitate was filtered, washed with water, and recrystallized from hexane.
2-Phenyl-6-(trifluoromethyl)-4H-pyran-4-one (5a). Method B. Yield 115 mg (48%), pale-yellow fine crystals, mp 85–86 °C. Procedure from dibromide 3a: Solutions of dibromide 3a (2.00 g, 5.0 mmol) in acetone (20 mL) was treated with Et3N (1.26 g, 12.5 mmol) under cooling on an ice bath. After that, the reaction mixture was stirred at room temperature for 3 days and diluted with H2O (75 mL). The precipitate was filtered, washed with water, and recrystallized from hexane. Yield 51% (0.60 g), pale-yellow fine crystals, mp 85–86 °C. IR (ATR) ν 3068, 1668, 1632, 1605, 1579, 1498, 1451, 1415. 1H NMR (400 MHz, CDCl3) δ 6.76 (1H, d, 4J = 2.1 Hz, CH); 6.84 (1H, d, 4J = 2.1 Hz, CH); 7.50–7.61 (3H, m, H Ph); 7.76–7.80 (m, 2H, H-2, H-6 Ph). 19F NMR (471 MHz, CDCl3) δ 90.3 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 112.3; 114.7 (q, 3JCF = 2.3 Hz, C-5); 118.5 (q, 1JCF = 274.8 Hz, CF3); 126.0 (2C Ar); 129.3 (2C Ar); 129.9; 132.2; 152.4 (q, 2JCF = 39.6 Hz, C-6); 163.9; 178.0 (CO). Anal. Calcd for C12H7F3O2: C 60.01; H 2.94. Found: C 59.63; H 2.56%.
2-(4-Fluorophenyl)-6-(trifluoromethyl)-4H-pyran-4-one (5b). Method A. Yield 0.18 g (69%), beige powder, mp 109–110 °C. IR (ATR) ν 3064, 1674, 1637, 1601, 1509, 1291, 1085, 945, 838. 1H NMR (400 MHz, CDCl3) δ 6.78 (1H, d, 4J = 2.1 Hz, CH); 6.82 (1H, d, 4J = 2.1 Hz, CH); 7.19–7.25 (2H, m, H-3, H-5 Ar); 7.76–7.85 (2H, m, H-2, H-6 Ar). 19F NMR (471 MHz, CDCl3) δ 55.8 (tt, 3JFH = 8.2 Hz, 4JFH = 5.1 Hz, F-Ar); 90.3 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 112.1 (d, 6JCF = 0.8 Hz, C-4); 114.8 (q, 3JCF = 2.6 Hz, C-5); 116.7 (d, 2JCF = 22.3 Hz, C-3, C-5 Ar); 118.3 (q, 1JCF = 273.5 Hz, CF3); 126.1 (d, 4JCF = 3.3 Hz, C-1 Ar); 128.3 (d, 3JCF = 9.0 Hz, C-2, C-6 Ar); 152.4 (q, 2JCF = 39.4 Hz, C-6); 163.0 (C-2); 164.9 (d, 1JCF = 253.2 Hz, C-4 Ar); 177.9 (CO). Anal. Calcd for C12H6F4O2·0.33H2O: C 54.57; H 2.54. Found: C 54.31; H 2.57%.
2-(4-Chlorophenyl)-6-(trifluoromethyl)-4H-pyran-4-one (5c). Method B. Yield 170 mg (62%), beige crystals, mp 135–137 °C. IR (ATR) ν 3061, 1671, 1632, 1605, 1492, 1416, 1407. 1H NMR (400 MHz, CDCl3) δ 6.76 (1H, d, 4J = 2.1 Hz, CH); 6.81 (1H, d, 4J = 2.1 Hz, CH); 7.51 (2H, d, 3J = 8.7 Hz, H Ar); 7.72 (2H, d, 3J = 8.7 Hz, H Ar). 19F NMR (376 MHz, CDCl3) δ 90.3 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 112.4 (C-3); 114.8 (q, 3JCF = 2.6 Hz, C-5); 118.4 (q, 1JCF = 273.7 Hz, CF3); 127.3 (2C Ar); 128.3 (C Ar); 129.7 (2C Ar); 138.7; 152.4 (q, 2JCF = 39.6 Hz, C-6); 162.8; 177.7 (CO). Anal. Calcd for C12H6ClF3O2: C 52.48; H 2.20. Found: C 52.47; H 2.40%.
2-(4-Bromophenyl)-6-(trifluoromethyl)-4H-pyran-4-one (5d). Method B. Yield 0.207 mg (65%), colorless powder, mp 161–163 °C. IR (ATR) ν 3059, 1668, 1654, 1645, 1627, 1600, 1590, 1488, 1414, 1402. 1H NMR (500 MHz, CDCl3) δ 6.76 (1H, d, 4J = 2.1 Hz, H-3); 6.82 (1H, d, 4J = 2.1 Hz, H-5); 7.64 (2H, d, 3J = 8.8 Hz, H Ar); 7.67 (2H, d, 3J = 8.8 Hz, H Ar). 19F NMR (471 MHz, CDCl3) δ 90.3 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 112.4 (C-3); 114.9 (q, 3JCF = 2.6 Hz, C-5); 118.4 (q, 1JCF = 273.9 Hz, CF3); 127.1 (C Ar); 127.4 (2C Ar); 128.8 (C Ar); 132.6 (2C Ar); 152.4 (q, 2JCF = 39.7 Hz, C-6); 162.9 (C-2); 177.7 (CO). Anal. Calcd for C12H6BrF3O2: C 45.17; H 1.90. Found: C 45.00; H 1.94%.
2-(p-Tolyl)-6-(trifluoromethyl)-4H-pyran-4-one (5f). Method B. Yield 71 mg (28%), colorless needles, 105–107 °C. IR (ATR) ν 1645, 1632, 1608, 1594, 1569, 1511, 1447. 1H NMR (500 MHz, CDCl3) δ 2.44 (3H, s, Me); 6.74 (1H, d, J = 2.1 Hz, H-5); 6.80 (1H, d, J = 2.1 Hz, H-3); 7.32 (2H, d, J = 8.1 Hz, H-3, H-5 Ar); 7.67 (2H, d, J = 8.1 Hz, H-2, H-6 Ar). 19F NMR (471 MHz, CDCl3) δ 90.3 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 21.5 (CH3); 111.6 (C-3); 114.6 (q, 3JCF = 2.5 Hz, C-5); 118.5 (q, 1JCF = 273.8 Hz, CF3); 125.9 (2C Ar); 127.0 (C Ar); 130.0 (2C Ar); 143.1(C Ar); 152.3 (q, 2JCF = 39.5 Hz, C-6); 164.1 (C-2); 178.0 (CO). HRMS (ESI) m/z [M + H]+. Calcd for C13H10F3O2: 255.0633. Found: 255.0625.
2-(4-Methoxyphenyl)-6-(trifluoromethyl)-4H-pyran-4-one (5g). Method A. Yield 176 mg (65%), yellow crystals, mp 106–107 °C. IR (ATR) ν 3019, 1685, 1598, 1522, 1489, 1432. 1H NMR (500 MHz, CDCl3) δ 3.89 (3H, s, Me); 6.73 (1H, d, 4J = 2.1 Hz, CH); 6.75 (1H, d, 4J = 2.1 Hz, CH); 7.02 (2H, d, 3J = 8.9 Hz, H-3, H-5 Ar); 7.73 (2H, d, 3J = 8.9 Hz, H-2, H-6 Ar). 19F NMR (376 MHz, CDCl3) δ 90.3 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 55.5 (CH3); 110.7 (C-3); 114.6 (q, 3JCF = 2.5 Hz, C-5); 114.7 (2C Ar); 118.6 (q, 1JCF = 273.6 Hz, CF3); 122.1 (C Ar); 127.8 (2C Ar); 152.1 (q, 2JCF = 39.4 Hz, C-6); 162.9; 164.0; 178.0 (CO). Anal. Calcd for C13H9F3O3: C 57.79; H 3.36. Found: C 57.39; H 3.39%.
2-(Thiophen-2-yl)-6-(trifluoromethyl)-4H-pyran-4-one (5i). Method A. Yield 153 mg (62%), yellow crystals, mp 87–88 °C. IR (ATR) ν 3042, 1668, 1652, 1630, 1594, 1427, 1408. 1H NMR (400 MHz, CDCl3) δ 6.68 (1H, d, J = 2.1 Hz, H-5); 6.71 (1H, d, J = 2.1 Hz, H-3); 7.19 (1H, dd, 3J = 4.9 Hz, 3J = 3.9 Hz, H-4 Th); 7.60 (1H, d, 3J = 4.9 Hz, H-5 Th); 7.65 (1H, d, 3J = 3.9 Hz, H-3 Th). 19F NMR (376 MHz, CDCl3) δ 90.3 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 110.7 (C-3); 114.8 (q, 3JCF = 2.4 Hz, C-5); 118.4 (q, 1JCF = 274.2 Hz, CF3); 128.7 (C Ar); 129.1 (C Ar); 130.9 (C Ar); 132.9 (C Ar); 151.9 (q, 2JCF = 39.7 Hz, C-6); 159.5; 177.4 (CO). HRMS (ESI) m/z [M + H]+. Calcd for C10H6F3O2S: 247.0041. Found: 247.0031.
2-(Difluoromethyl)-6-phenyl-4H-pyran-4-one (5j). Yield 40% (89 mg), yellow powder, mp 135–136 °C. IR (ATR) ν 1665, 1616, 1596, 1497, 1358, 1177, 1108, 1055, 925. 1H NMR (500 MHz, CDCl3) δ 6.47 (1H, t, 2J = 53.7 Hz, CHF2); 6.63 (1H, d, 4J = 2.2 Hz, H-3); 6.84 (1H, d, 4J = 2.2 Hz, H-5); 7.49–7.58 (3H, m, H Ph); 7.78 (2H, dd, 3J = 8.1 Hz, 4J = 1.1 Hz, H-2, H-6 Ph). 19F NMR (471 MHz, CDCl3) δ 38.3 (d, 2J = 53.7 Hz, CF2H). 13C NMR (126 MHz, CDCl3) δ 109.0 (t, 1JCF = 242.9 Hz, CF2H); 112.2; 114.4 (t, 3JCF = 4.0 Hz, C-3); 126.0 (2C Ar); 129.2 (2C Ar); 130.3; 132.0; 157.0 (t, 2JCF = 27.2 Hz, C-2); 163.9; 178.6 (CO). HRMS (ESI) m/z [M + H]+. Calcd for C12H9F2O2: 223.0571. Found: 223.0574.
2-Phenyl-6-(1,1,2,2-tetrafluoroethyl)-4H-pyran-4-one (5k). Yield 44% (120 mg), yellow powder, mp 125–126 °C. IR (ATR) ν 3074, 2924, 1663, 1622, 1452, 1239, 1104, 940. 1H NMR (500 MHz, CDCl3) δ 6.10 (1H, tt, 2J = 53.1 Hz, 3J = 2.7 Hz, CHF2); 6.77 (1H, d, 4J = 1.8 Hz, CH); 6.84 (1H, d, 4J = 1.8 Hz, CH); 7.50–7.60 (3H, m, H Ph); 7.76 (2H, dd, 3J = 7.8 Hz, 4J = 1.4 Hz, H-2, H-6 Ph). 19F NMR (471 MHz, CDCl3) δ 26.9 (dt, 2JHF = 53.0 Hz, 3JFF = 4.0 Hz, CF2); 41.1 (td, 3JFF = 4.0 Hz, 3JHF = 2.7 Hz, CF2H). HRMS (ESI) m/z [M + H]+. Calcd for C13H9F4O2: 273.0539. Found: 273.0542.
2-(1,1,2,2,3,3,4,4-Octafluorobutyl)-6-phenyl-4H-pyran-4-one (5l). Yield 70% (261 mg), pale-yellow semi-solid liquid. IR (ATR) ν 3047, 1660, 1629, 1602, 1497, 1404, 1118, 944, 767. 1H NMR (500 MHz, CDCl3) δ 6.08 (1H, tt, 2J = 51.8 Hz, 3J = 5.1 Hz, CHF2); 6.83 (1H, d, 4J = 2.0 Hz, CH); 6.88 (1H, d, 4J = 2.0 Hz, CH); 7.51 (2H, dd, 3J = 7.9 Hz, 3J = 7.3 Hz, H-3, H-5 Ph); 7.57 (1H, tt, 3J = 7.3 Hz, 4J = 1.3 Hz, H-4 Ph); 7.75 (2H, dd, 3J = 7.9 Hz, 4J = 1.3 Hz, H-2, H-6 Ph). 19F NMR (376 MHz, CDCl3) δ 24.8 (dm, 2JHF = 51.8 Hz, CF2H); 32.5–32.7 (m, 3-CF2); 37.75–37.85 (m, 2-CF2); 43.8 (tt, 3JFF = 11.4 Hz, 3JFF = 1.7 Hz, 1-CF2). HRMS (ESI) m/z [M + H]+. Calcd for C15H9F8O2: 373.0475. Found: 373.0479.
3.6. Synthesis of Pyrones 6
General procedure (Method C). A solution of Br2 (400 mg, 2.5 mmol) in CH2Cl2 (4.25 mL) was added to enedione 2 (1 mmol) in CH2Cl2 (2.7 mL) under cooling on an ice-bath. After that, the reaction mixture was stirred at room temperature for 15 h, and then the solvent was evaporated without heating. The residue was refluxed in pyridine (3 mL) at 100 °C for 10 h. The excess of pyridine was evaporated under reduced pressure and the product was purified by flash-chromatography (EtOAc). The product was hot-extracted with hexane (3 × 5 mL). The solvent was evaporated until 2 mL of hexane, and the product crystallized from the extract after cooling.
3-Bromo-6-phenyl-2-(trifluoromethyl)-4H-pyran-4-one (6a). Yield 73 mg (23%), beige solid, mp 132–133 °C. IR (ATR) ν 3069, 1646, 1600, 1451, 1146, 973, 769. 1H NMR (500 MHz, CDCl3) δ 6.94 (1H, s, H-5); 7.50–7.57 (2H, m, H-3, H-5 Ph); 7.57–7.62 (1H, m, H-4 Ph); 7.79 (2H, d, 3J = 7.4 Hz, H-2, H-6 Ph). 19F NMR (471 MHz, CDCl3) δ 95.6 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 109.3; 116.3; 118.8 (q, 1JCF = 276.4 Hz, CF3); 126.0 (2C Ph); 129.3; 129.4 (2C Ph); 132.6; 149.7 (q, 2JCF = 38.0 Hz, C-6); 163.2; 172.8 (CO). Anal. Calcd for C12H6BrF3O2: C 45.17; H 1.90. Found: C 45.36; H 1.86%.
3-Bromo-6-(4-fluorophenyl)-2-(trifluoromethyl)-4H-pyran-4-one (6b). Yield 94 mg (28%), beige solid, mp 156–167 °C. IR (ATR) ν 1667, 1627, 1556, 1412. 1H NMR (500 MHz, CDCl3) δ 6.88 (1H, s, H-5); 7.20–7.28 (2H, m, H-3, H-5 Ar); 7.77–7.85 (2H, m, H-2, H-6 Ar). 19F NMR (471 MHz, CDCl3) δ 56.4 (tt, 3JFH = 8.2 Hz, 4JFH = 5.2 Hz, F-Ar); 95.6 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 109.1 (d, 6JCF = 1.1 Hz, H-5); 116.4 (q, 3JCF = 1.1 Hz, C-3); 116.8 (d, 2JCF = 22.3 Hz, C-3, C-5 Ar); 118.7 (q, 1JCF = 276.6 Hz, CF3); 125.5 (d, 4JCF = 3.4 Hz, C-1 Ar); 128.4 (d, 3JCF = 9.1 Hz, C-2, C-6 Ar); 149.6 (q, 2JCF = 39.6 Hz, C-2); 162.2; 162.3 (d, 1JCF = 255.4 Hz, C-4 Ar); 172.7 (CO). Anal. Calcd for C12H5BrF4O2: C 42.76; H 1.50. Found: C 42.39; H 1.33%.
3-Bromo-6-(4-chlorophenyl)-2-(trifluoromethyl)-4H-pyran-4-one (6c). Yield 120 mg (34%), beige solid, mp 149–150 °C. Procedure from tribromide 3c: Tribromide 3c (533 mg, 1.0 mmol) was refluxed in pyridine (3 mL) at 100 °C for 10 h. The excess of pyridine was evaporated under reduced pressure, and the product purified by flash-chromatography (EtOAc). The product was hot-extracted with hexane (3×5 mL). The solvent was evaporated until 2 mL of hexane, and the product crystallized from extract after cooling. Yield 36% (127 mg), beige solid, mp 149–150 °C. IR (ATR) ν 3069, 1645, 1600, 1577, 1561, 1498, 1451. 1H NMR (400 MHz, CDCl3) δ 6.91 (1H, s, H-5); 7.52 (2H, d, 3J = 8.7 Hz, H Ar); 7.72 (2H, d, 3J = 8.7 Hz, H Ar). 19F NMR (376 MHz, CDCl3) δ 95.6 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 109.4 (C-5); 116.5 (C-3); 118.7 (q, 1JCF = 276.5 Hz, CF3); 127.3 (2C Ar); 127.7 (C Ar); 129.8 (2C Ar); 139.1; 149.7 (q, 2JCF = 38.1 Hz, C-2); 162.1; 172.7 (CO). HRMS (ESI) m/z [M + H]+. Calcd for C12H6BrClF3O2: 352.9186. Found: 352.9183.
3-Bromo-6-(3-nitrophenyl)-2-(trifluoromethyl)-4H-pyran-4-one (6e). Yield 15 mg (4%), yellowish solid, mp 122–125 °C. IR (ATR) ν 3074, 3055, 1672, 1647, 1615, 1596, 1532, 1442. 1H NMR (500 MHz, DMSO-d6) δ 7.58 (1H, s, H-5); 7.90 (1H, t, 3J = 8.1 Hz, H-5 Ar); 8.39 (1H, ddd, 3J = 7.9 Hz, 4J = 1.7 Hz, 4J = 0.9 Hz, H-6 Ar); 8.46 (1H, ddd, 3J = 8.2 Hz, 4J = 2.2 Hz, 4J = 0.8 Hz, H-4 Ar); 8.68 (1H, dd, 4J = 2.2 Hz, 4J = 1.7 Hz, H-2 Ar). 1H NMR (400 MHz, CDCl3) δ 7.04 (1H, s, H-5); 7.78 (1H, t, 3J = 8.8 Hz, H-5 Ar); 8.12 (1H, ddd, 3J = 7.9 Hz, 4J = 1.7 Hz, 4J = 1.0 Hz, H-6 Ar); 8.45 (1H, ddd, 3J = 8.3 Hz, 4J = 2.2 Hz, 4J = 1.0 Hz, H-4 Ar); 8.64 (1H, dd, 4J = 2.2 Hz, 4J = 1.7 Hz, H-2 Ar). 19F NMR (376 MHz, CDCl3) δ 95.6 (s, CF3). 13C NMR (126 MHz, DMSO-d6) δ 110.9; 115.8; 118.6 (q, 1JCF = 277.0 Hz, CF3); 120.8; 126.5; 131.0; 131.2; 132.4; 148.3; 148.9 (q, 2JCF = 36.8 Hz, C-2); 160.2; 172.5 (CO). HRMS (ESI) m/z [M + H]+. Calcd for C12H6BrF3NO4: 363.9427. Found: 363.9429.
3-Bromo-6-(p-tolyl)-2-(trifluoromethyl)-4H-pyran-4-one (6f). Yield 107 mg (32%), beige solid, mp 170–171 °C. IR (ATR) ν 1644, 1608, 1595, 1569, 1511, 1449. 1H NMR (500 MHz, CDCl3) δ 2.44 (3H, s, Me); 6.90 (1H, s, H-5); 7.33 (2H, d, J = 8.1 Hz, H-3, H-5 Ar); 7.67 (2H, d, J = 8.1 Hz, H-2, H-6 Ar). 19F NMR (471 MHz, CDCl3) δ 95.6 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 21.5 (CH3); 111.6 (C-3); 114.6 (q, 3JCF = 2.5 Hz, C-5); 118.5 (q, 1JCF = 273.8 Hz, CF3); 125.9 (2C Ar); 127.0 (C Ar); 130.0 (2C Ar); 143.1(C Ar); 152.3 (q, 2JCF = 39.5 Hz, C-6); 164.1 (C-2); 178.0 (CO). HRMS (ESI) m/z [M + H]+. Calcd for C13H9BrF3O2: 332.9733. Found: 332.9737.
3-Bromo-2-(difluoromethyl)-6-phenyl-4H-pyran-4-one (6j). Yield 84 mg (28%), beige solid, mp 122–123 °C. 1H NMR (500 MHz, CDCl3) δ 6.93 (1H, s, H-5); 7.00 (1H, t, 2JHF = 52.1 Hz, CHF2); 7.50–7.61 (3H, m, H Ph); 7.80–7.84 (2H, m, H-2, H-6 Ph). 19F NMR (376 MHz, CDCl3) δ 39.3 (d, 2JHF = 52.1 Hz, CF2H). 13C NMR (126 MHz, CDCl3) δ 109.3 (t, 1JCF = 242.3 Hz, CF2H); 109.5; 116.5 (t, 3JCF = 5.3 Hz, C-3); 126.1 (2C Ph); 129.3 (2C Ph); 129.6; 132.4; 152.9 (t, 2JCF = 23.9 Hz, C-2); 163.6; 172.9 (CO). HRMS (ESI) m/z [M + H]+. Calcd for C13H10F3O2: 255.0633. Found: 255.0629.
3-Bromo-6-phenyl-2-(1,1,2,2-tetrafluoroethyl)-4H-pyran-4-one (6k). Yield 77 mg (24%), beige solid, mp 122–125 °C. IR (ATR) ν 3074, 3002, 1642, 1452, 1383, 1231, 1213, 1083, 975, 842. 1H NMR (500 MHz, CDCl3) δ 6.32 (1H, tt, 2JHF = 52.8 Hz, J = 4.0 Hz, CHF2); 6.95 (1H, s, H-5); 7.51–7.56 (2H, m, H-3, H-5 Ph); 7.57–7.61 (1H, m, H-4 Ph); 7.75–7.79 (2H, m, H-2, H-6 Ph). 19F NMR (471 MHz, CDCl3) δ 26.3 (dt, 3JFF = 6.6 Hz, 3JHF = 4.1 Hz, CF2); 44.6 (td, 2JFF = 52.8 Hz, 3JFF = 6.6 Hz, CF2H). 13C NMR (126 MHz, CDCl3) δ 108.9 (tt, 1JCF = 254.1 Hz, 2JCF = 35.8 Hz, CF2H); 109.2; 111.4 (tt, 1JCF = 256.9 Hz, 2JCF = 28.9 Hz, CF2); 117.6; 126.0 (2C Ph); 129.4 (2C Ph); 130.5; 132.5; 150.9 (t, 2JCF = 27.4 Hz, C-2); 163.6; 172.8 (CO). HRMS (ESI) m/z [M + H]+. Calcd for C13H7BrF4O2: 350.9644. Found: 350.9650.
3-Bromo-2-(1,1,2,2,3,3,4,4-octafluorobutyl)-6-phenyl-4H-pyran-4-one (6l). Yield 149 mg (33%), beige solid, mp 86–88 °C. IR (ATR) ν 3075, 1640, 1499, 1453, 1377, 1145, 943. 1H NMR (400 MHz, CDCl3) δ 6.12 (3H, tt, 2JHF = 51.9 Hz, 3JHF = 5.2 Hz, CHF2); 6.95 (1H, s, H-5); 7.51–7.62 (3H, m, H Ph); 7.74–7.78 (2H, m, H-2, H-6 Ph). 19F NMR (376 MHz, CDCl3) δ 24.9 (dm, 2JHF = 51.9 Hz, CF2H); 32.4 –32.6 (m, 3-CF2); 38.7–38.8 (m, 2-CF2); 49.3 (t, 3JFF = 11.6 Hz, 1-CF2). HRMS (ESI) m/z [M + H]+. Calcd for C15H7BrF8O2: 450.9580. Found: 450.9590.
3.7. Synthesis of Fluorinated Azaheterocycles 7–9
2-Phenyl-6-(trifluoromethyl)pyridin-4(1H)-one (7). A solution of pyrone 5a (120 mg, 0.5 mmol) in EtOH (2 mL), saturated with NH3 at room temperature, was heated in an autoclave at 100 °C for 8 h. The reaction mixture was diluted with H2O (10 mL), and the formed precipitate was filtered and recrystallized from EtOH. Yield 84 mg (70%), colorless crystals, mp 133–134 °C. IR (ATR) ν 1614, 1579, 1481, 1427, 1460, 1429. 1H NMR (400 MHz, DMSO-d6) δ pyridinol form (96%): 7.05 (1H, s, H-3); 7.25 (1H, s, H-5); 7.39–7.48 (3H, m, H Ph); 7.91 (2H, d, J = 6.8 Hz, H-2, H-6 Ph); pyridone form (4%): 6.87 (1H, d, 4J = 2.0 Hz, H-3); 6.84 (1H, d, 4J = 2.0 Hz, H-5); 7.39–7.48 (3H, m, H Ph); 7.87–7.94 (2H, m, H-2, H-6 Ph); the signals from NH and OH were not observed due to the fast exchange with water. 19F NMR (376 MHz, CDCl3) δ pyridone form (4%): 93.4 (s, CF3); pyridinol form (96%): 93.6 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 108.3 Hz, 111.2 Hz, 121.2 (q, 1JCF = 274.4 Hz, CF3); 127.1 (2C Ph); 128.9 (2C Ph); 130.0 Hz, 137.2 Hz, 148.1 Hz, 158.9 Hz, 166.5. Anal. Calcd for C12H8F3NO: C 60.26; H 3.37; N 5.86. Found: C 60.35; H 3.26; N 5.84%.
1-Phenyl-3-(4-(trifluoromethyl)-1H-1,2,3-triazol-5-yl)propane-1,3-dione (8). To a solution of pyrone 5a (96 mg, 0.4 mmol) in DMSO (1 mL), NaN3 (29 mg, 0.44 mmol) was added, and the mixture was stirred at 115–120 °C for 30 min. The reaction was then diluted with aqueous HCl (10 mL, 0.1 M), and the formed precipitate was filtered and recrystallized from toluene. Yield 65 mg (55%), pale-brown plates, mp 189–190 °C. IR (ATR) ν 3268 (NH), 1600, 1575, 1535, 1492, 1460, 1429. 1H NMR (400 MHz, DMSO-d6) δ enol form (82%): 7.19 (1H, s, 2-CH); 7.59 (2H, t, J = 7.6 Hz, H-3, H-5 Ph); 7.68 (1H, t, J = 7.4 Hz, H-4 Ph); 8.05 (2H, d, J = 7.4 Hz, H-2, H-6 Ph); 14.70–17.9 (2H, broad s, NH, OH); keto form (18%): 4.85 (2H, s, CH2); 7.58 (2H, t, J = 7.4 Hz, H-3, H-5 Ph); 7.70 (1H, t, J = 7.4 Hz, H-4 Ph); 8.01 (2H, d, J = 7.4 Hz, H-2, H-6 Ph); 14.70–17.9 (1H, broad s, NH). 19F NMR (376 MHz, DMSO-d6) δ keto form (18%): 102.2 (s, CF3); enol form (82%): 103.2 (s, CF3). 13C NMR (126 MHz, DMSO-d6) δ enol form: 94.9 (CH=); 120.7 (q, 1JCF = 268.5 Hz, CF3); 127.2 (2C Ar); 129.1 (2C Ar); 133.2 (C Ar); 133.2 (C Ar); 136.2 (q, 2JCF = 40.1 Hz, C-CF3); 140.9 (C Ar); 179.6; 182.2; keto form: 50.7 (CH2); 120.2 (q, 1JCF = 267.1 Hz, CF3); 128.5 (2C Ar); 128.9 (2C Ar); 133.9 (C Ar); 136.0 (C Ar); 136.2 (q, 2JCF = 40.5 Hz, C-CF3); 142.4 (C Ar); 187.9 (CO); 194.8 (CO). Anal. Calcd for C12H8F3N3O2: C 50.89; H 2.85; N 14.84. Found: C 50.90; H 2.78; N 14.85%.
(E)-1,5-Diphenyl-3-(3,3,3-trifluoro-2-(2-phenylhydrazono)propyl)-1H-pyrazole (9). A mixture of pyrone 5a (120 mg, 0.5 mmol) and phenylhydrazine (270 mg, 2.5 mmol) was heated at 120 °C for 3 h. The reaction mixture was cooled to room temperature, treated with 1M HCl solution (10 mL), the formed precipitate was filtered and recrystallized from EtOH. Yield 65 mg (55%), colorless crystals, mp 171–173 °C. IR (ATR) ν 3267 (NH), 1605 (C=N), 1543, 1500, 1457. 1H NMR (500 MHz, CDCl3) δ 3.84 (2H, s, H-3); 6.40 (1H, s, H-5); 6.91 (1H, t, J = 7.3 Hz, H-4 Ph); 7.11 (2H, d, J = 7.8 Hz, H-2, H-6 Ph); 7.19 (2H, dd, 3J = 7.7 Hz, 4J = 1.5 Hz, H-2, H-6 Ph); 7.22–7.38 (3H, m, H Ph); 9.99 (1H, s, NH). 19F NMR (376 MHz, CDCl3) δ 92.8 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 24.6; 107.2; 113.5 (2C); 121.4; 121.94 (q, 1JCF = 272.0 Hz, CF3); 125.1 (2C); 127.8; 128.5 (2C); 128.6; 128.7 (2C); 128.78 (q, 2JCF = 33.9 Hz, C=N); 129.0 (2C); 129.2 (2C); 129.8; 139.6; 144.1; 144.8; 147.1. Anal. Calcd for C24H19F3N4: C 68.56; H 4.56; N 13.33. Found: C 68.42; H 4.38; N 13.34%.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
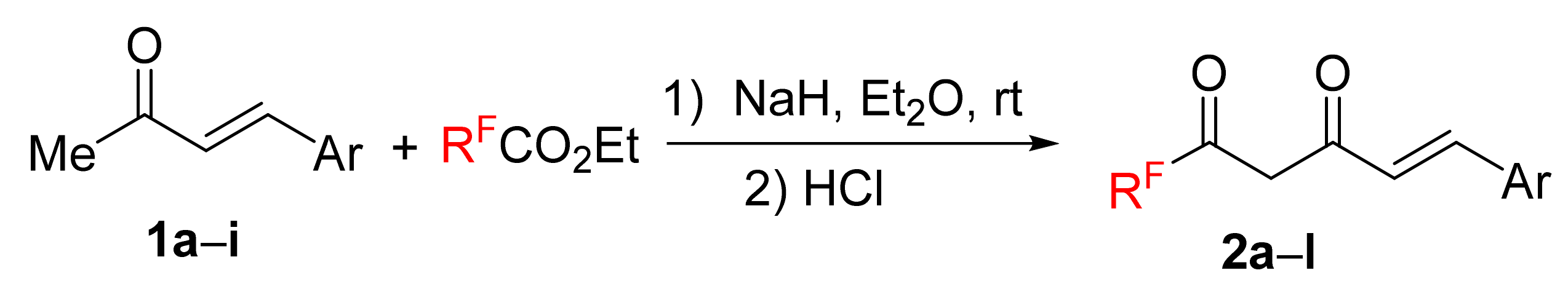{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}