
Inhibition of Cysteine Proteases by 6,6′-Dihydroxythiobinupharidine (DTBN) from Nuphar lutea

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Computaitonal Analyses
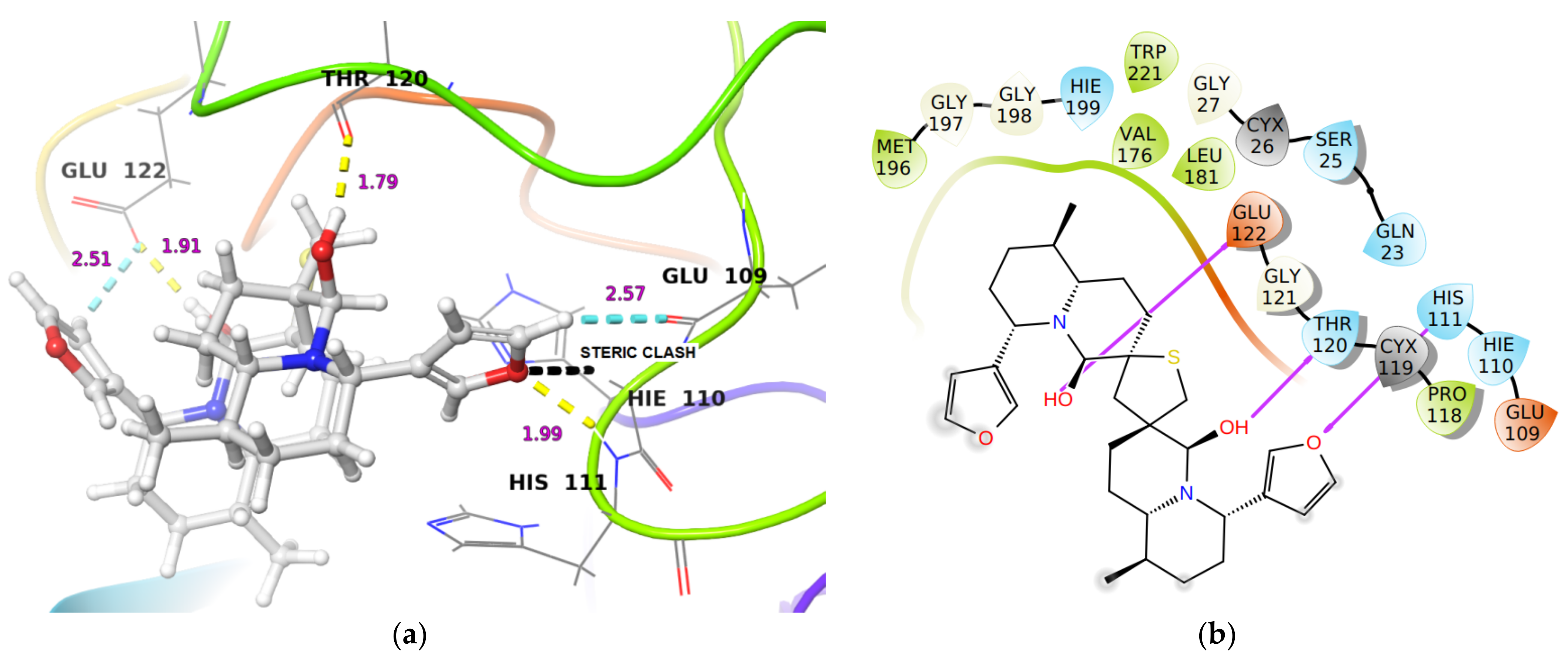
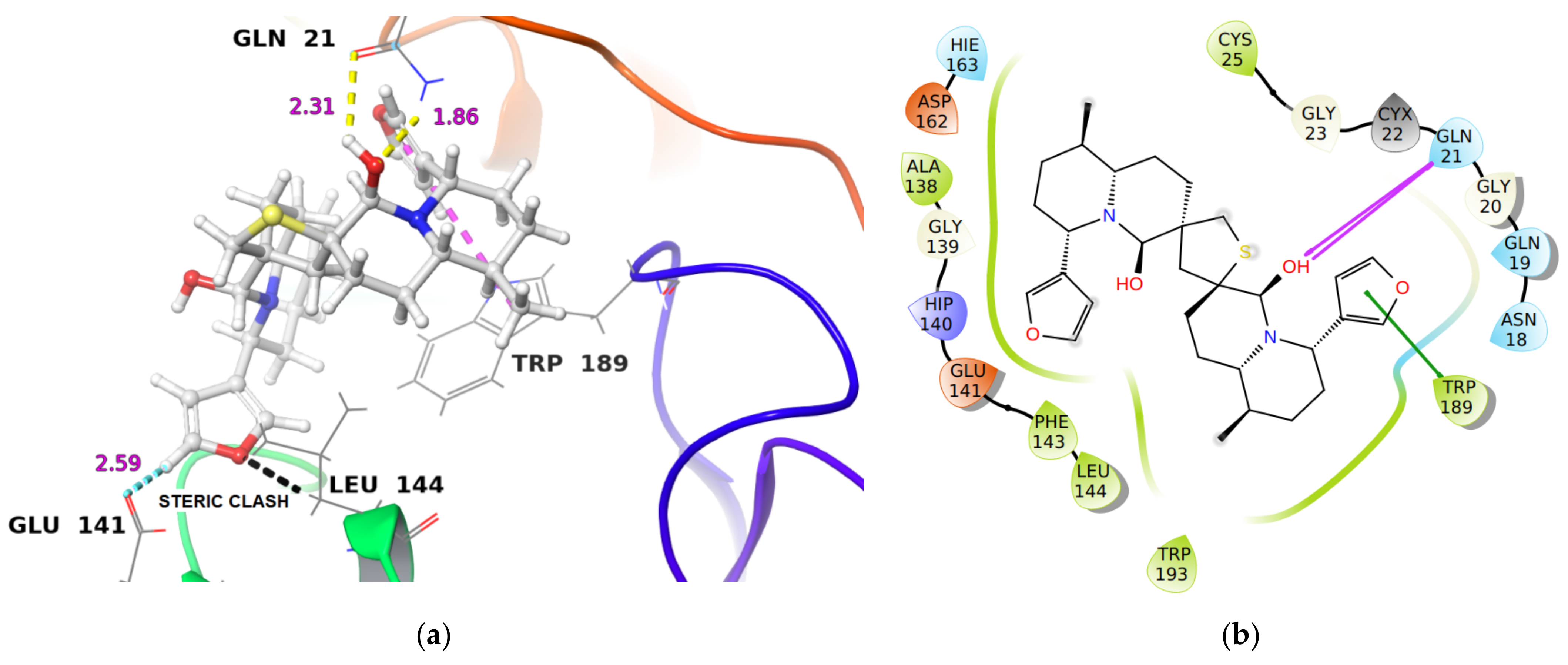
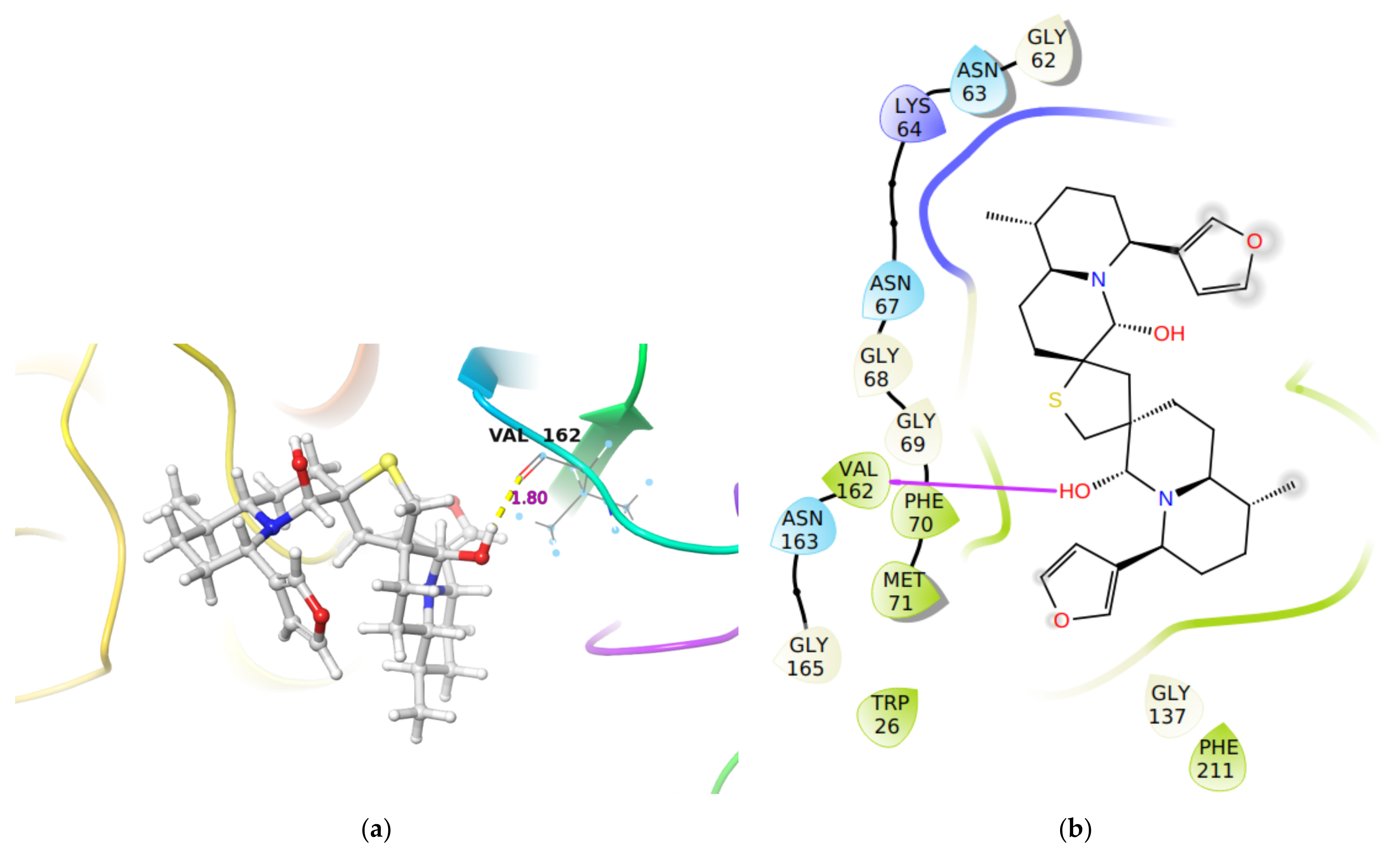
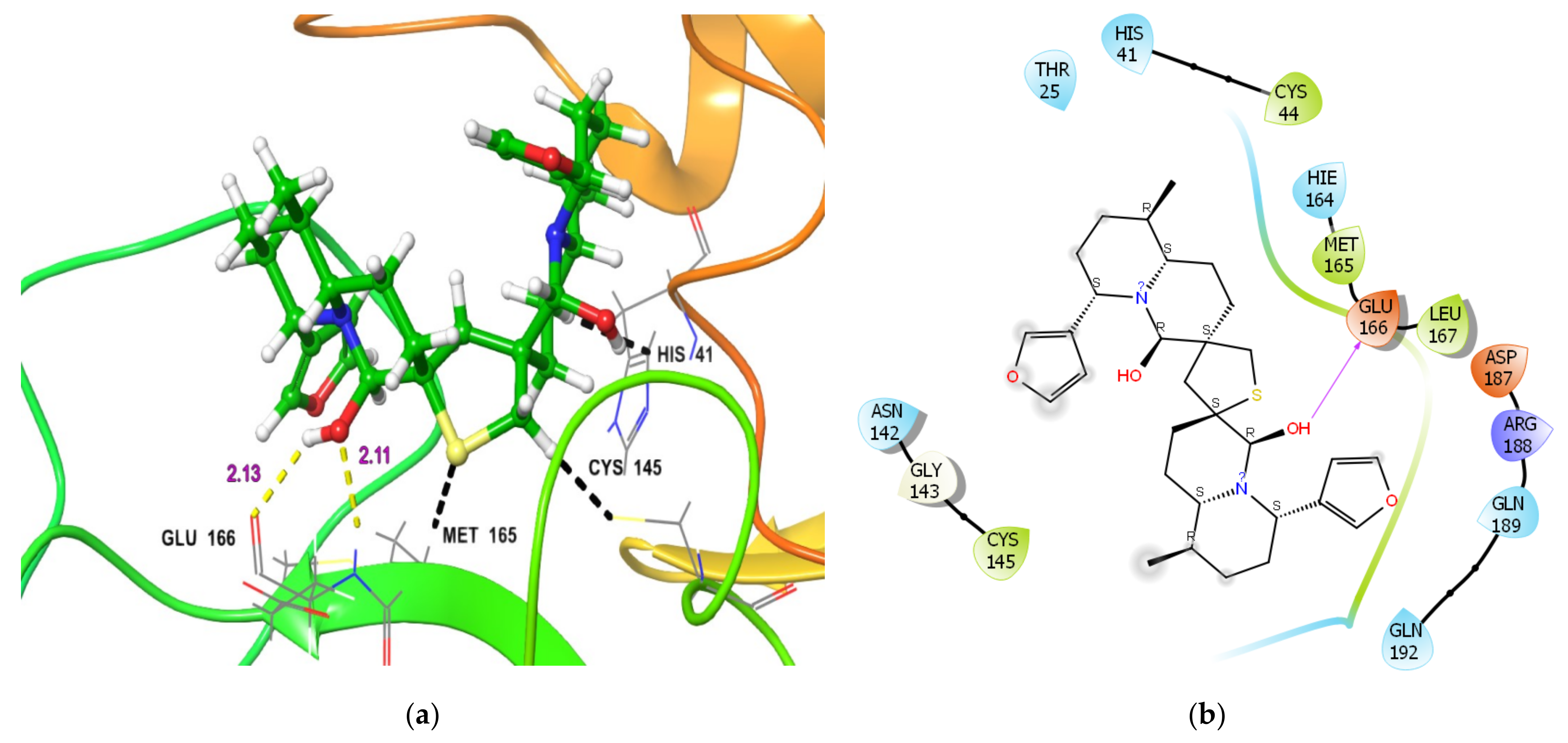
2.1.1. Molecular Docking Analysis
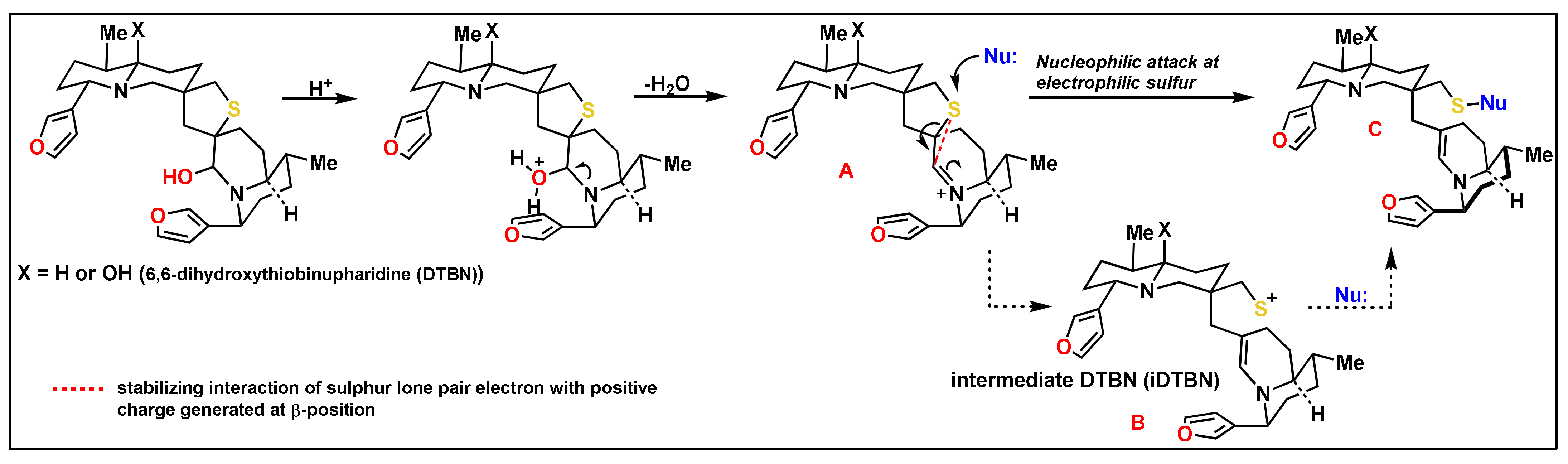
2.1.2. Covalent Docking Analysis
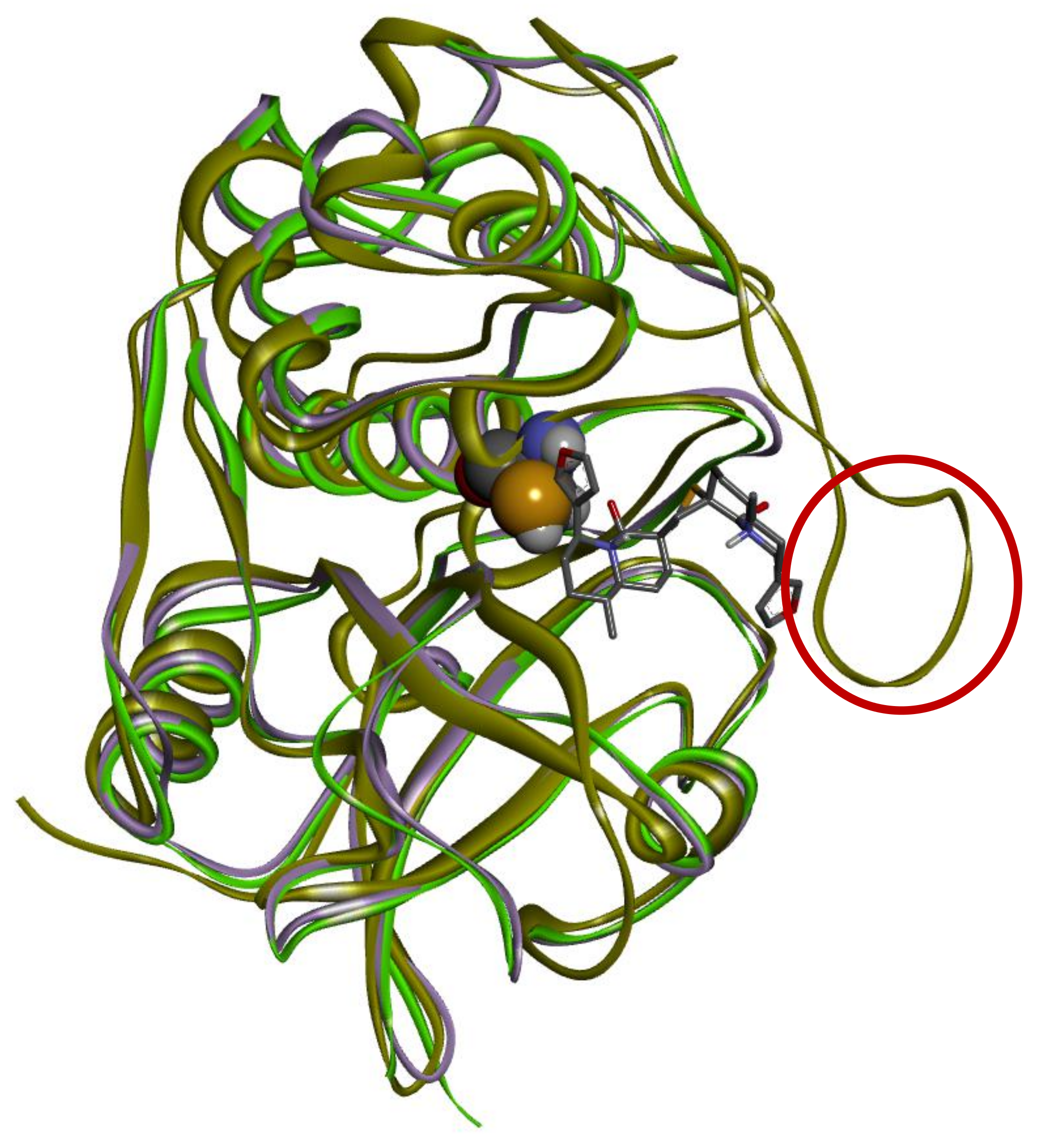
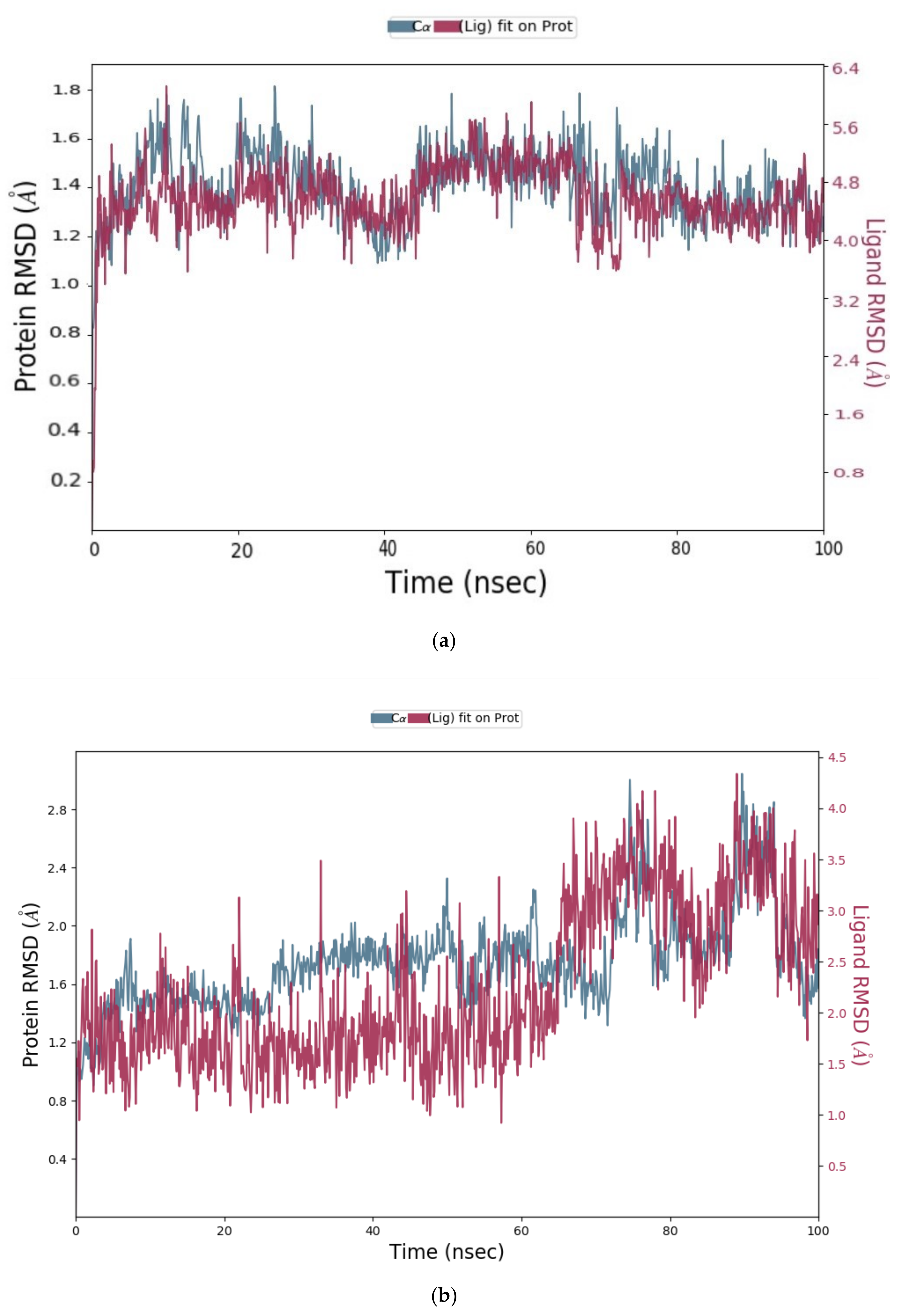
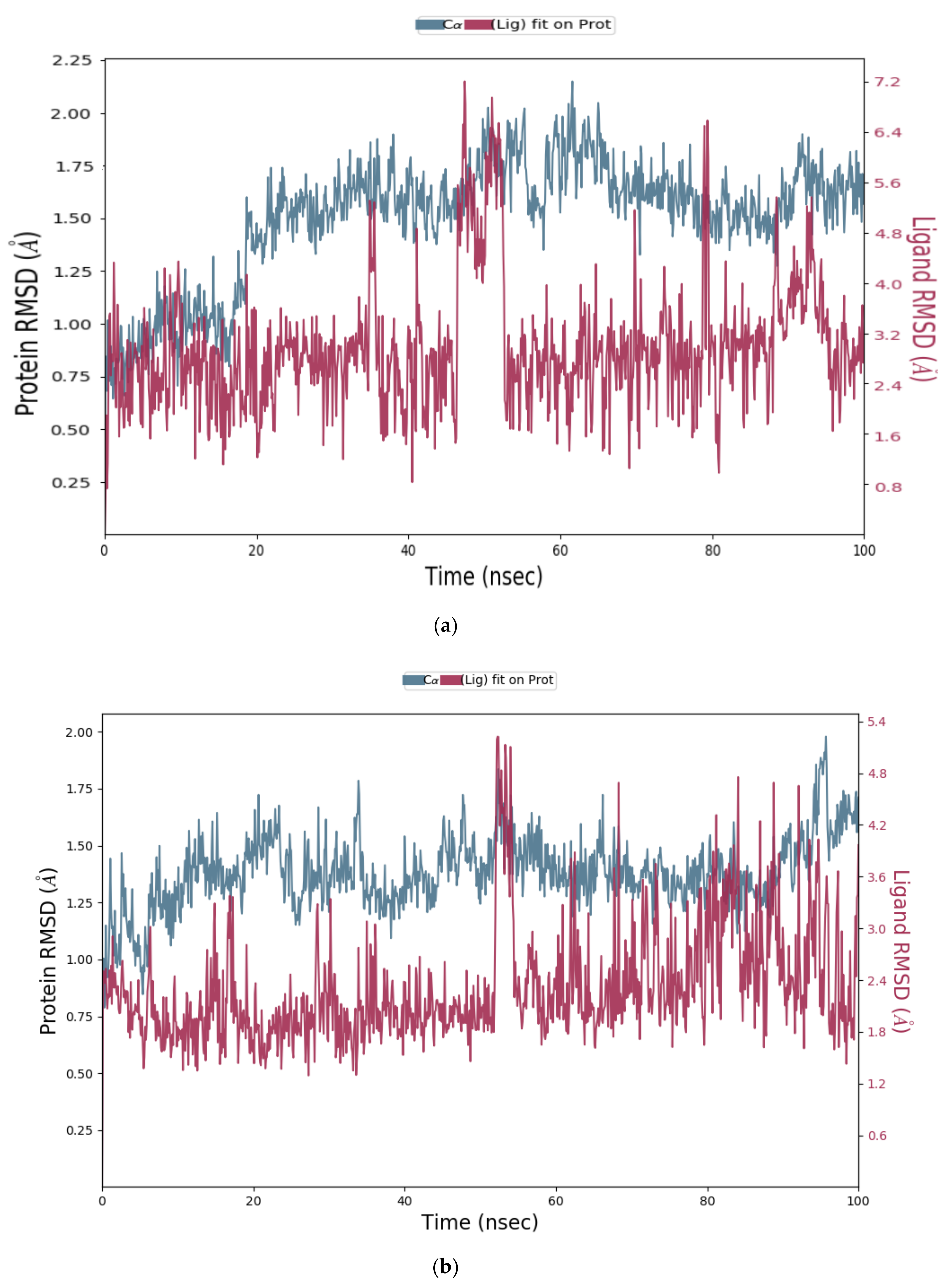
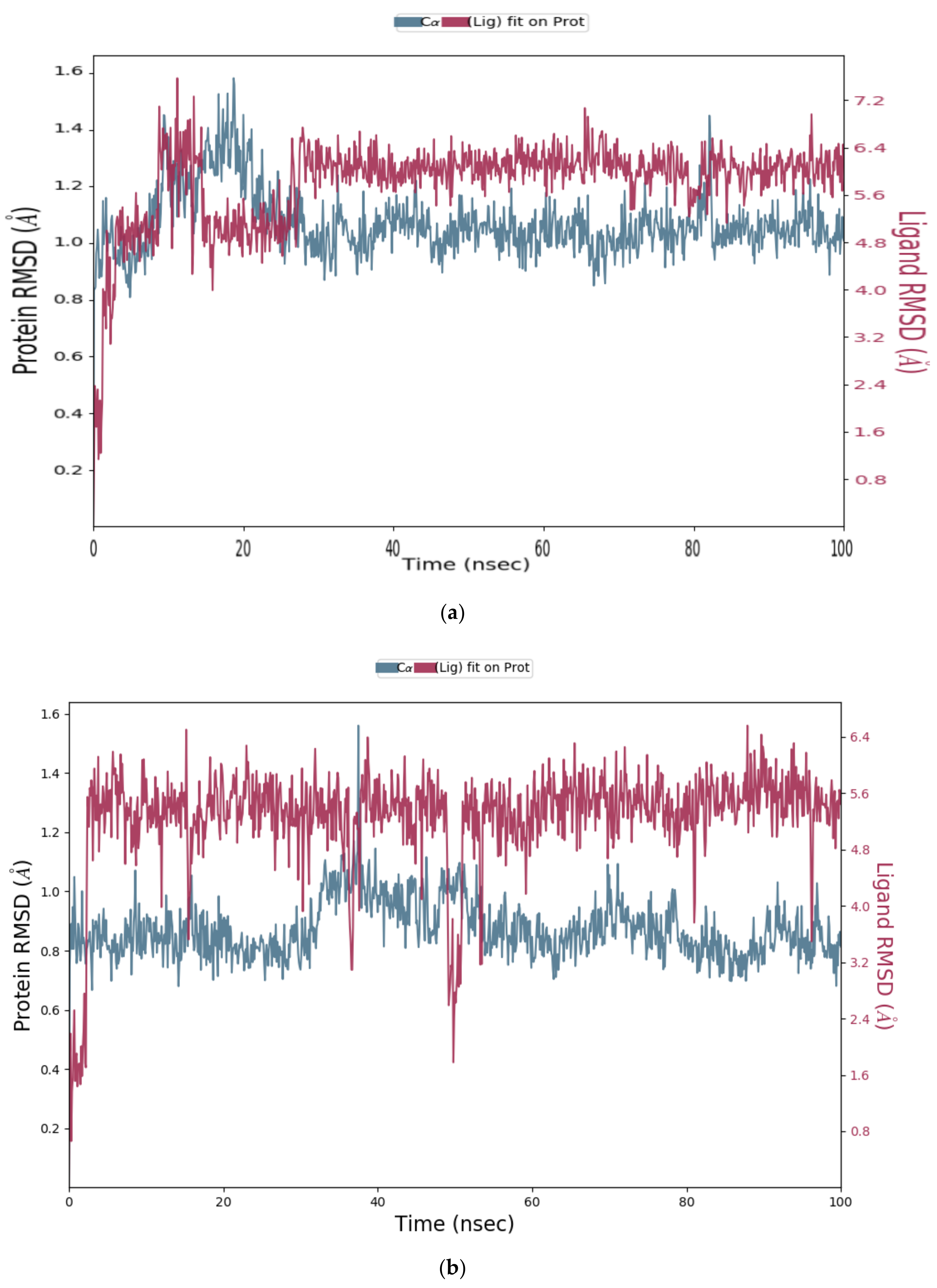
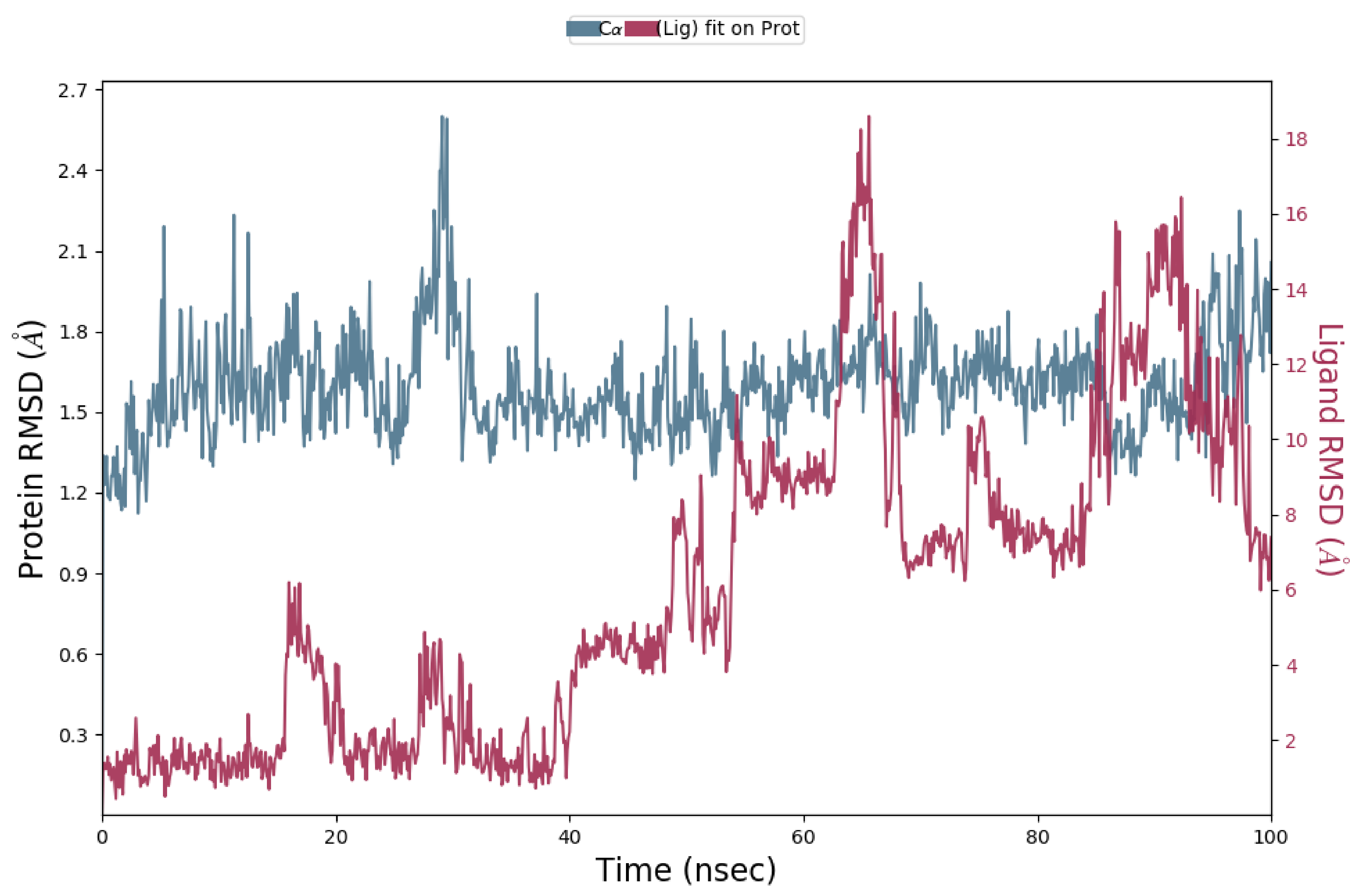
2.1.3. Molecular Dynamics (MD) Simulation
Analysis of RMSD Value of Proteins and Ligands
Analysis of RMSF Value of Proteins
Molecular Interactions Observed during MD Simulations
Molecular Interactions That Exist above 30% of Minimum Contact Strength
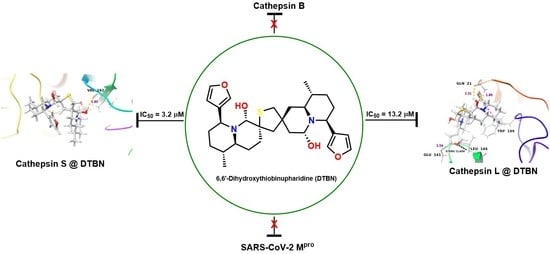
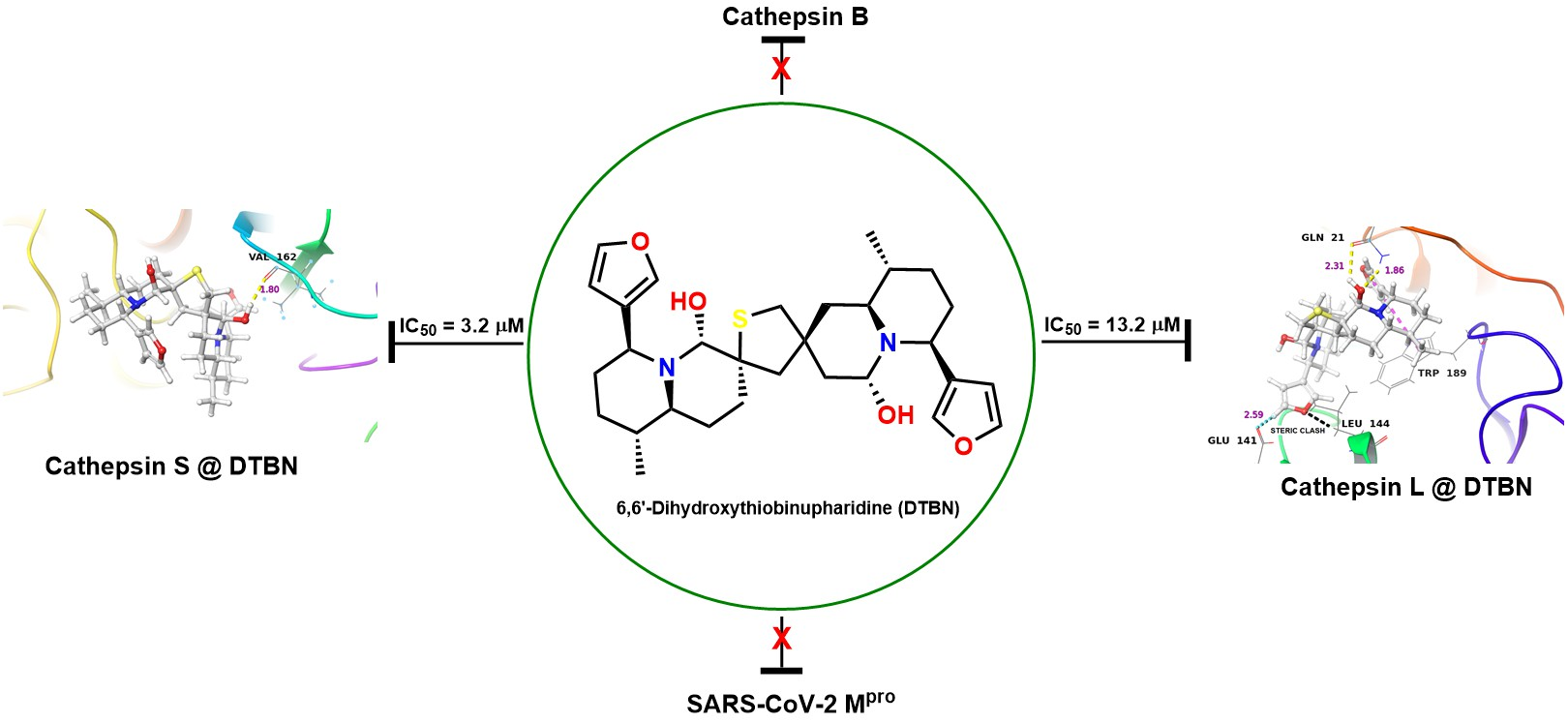
2.2. In Vitro Analysis
Enzyme Activities
3. Materials and Methods
3.1. Materials
3.2. Computational Methodology
3.2.1. Molecular Docking
3.2.2. Retrieval of Protein Crystal Structures
3.2.3. Ligand Preparation
3.2.4. Molecular Docking and MM-GBSA Refinement
3.2.5. Covalent Docking
3.2.6. Molecular Dynamics (MD) Simulation
3.3. In Vitro Methodology
Enzymes Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Yamahara, J.; Shimoda, H.; Matsuda, H.; Yoshikawa, M. Potent immunosuppressive principles, dimeric sesquiterpene thioalkaloids, isolated from nupharis rhizoma, the rhizoma of Nuphar pumilum (nymphaeaceae): Structure-requirement of nuphar-alkaloid for immunosuppressive activity. Biol. Pharm. Bull. 1996, 19, 1241–1243. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, H.; Morikawa, T.; Oda, M.; Asao, Y.; Yoshikawa, M. Potent anti-metastatic activity of dimeric sesquiterpene thioalkaloids from the rhizome of Nuphar pumilum. Bioorg. Med. Chem. Lett. 2003, 13, 4445–4449. [Google Scholar] [CrossRef]
- Matsuda, H.; Yoshida, K.; Miyagawa, K.; Nemoto, Y.; Asao, Y.; Yoshikawa, M. Nuphar alkaloids with immediately apoptosis-inducing activity from Nuphar pumilum and their structural requirements for the activity. Bioorg. Med. Chem. Lett. 2006, 16, 1567–1573. [Google Scholar] [CrossRef]
- Modzelewska, A.; Sur, S.; Kumar, S.K.; Khan, S.R. Sesquiterpenes: Natural products that decrease cancer growth. Curr. Med. Chem. Anticancer Agents 2005, 5, 477–499. [Google Scholar] [CrossRef] [PubMed]
- Ozer, J.; Eisner, N.; Ostrozhenkova, E.; Bacher, A.; Eisenreich, W.; Benharroch, D.; Golan-Goldhirsh, A.; Gopas, J. Nuphar lutea thioalkaloids inhibit the nuclear factor kappaB pathway, potentiate apoptosis and are synergistic with cisplatin and etoposide. Cancer Biol. Ther. 2009, 8, 1860–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozer, J.; Levi, T.; Golan-Goldhirsh, A.; Gopas, J. Anti-inflammatory effect of a Nuphar lutea partially purified leaf extract in murine models of septic shock. J. Ethnopharmacol. 2015, 161, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, A.B.; Karakas, F.P.; Turker, A.U. In vitro antibacterial and antitumor activities of some medicinal plant extracts, growing in Turkey. Asian Pac. J. Trop. Med. 2013, 6, 616–624. [Google Scholar] [CrossRef]
- Okamura, S.; Nishiyama, E.; Yamazaki, T.; Otsuka, N.; Taniguchi, S.; Ogawa, W.; Hatano, T.; Tsuchiya, T.; Kuroda, T. Action mechanism of 6, 6′-dihydroxythiobinupharidine from Nuphar japonicum, which showed anti-MRSA and anti-VRE activities. Biochim. Biophys. Acta 2015, 1850, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Cullen, W.P.; LaLonde, R.T.; Wang, C.J.; Wong, C.F. Isolation and in vitro antifungal activity of 6,6′-dihydroxythiobinupharidine. J. Pharm. Sci. 1973, 62, 826–827. [Google Scholar] [CrossRef]
- El-On, J.; Ozer, L.; Gopas, J.; Sneir, R.; Golan-Goldhirsh, A. Nuphar lutea: In vitro anti-leishmanial activity against Leishmania major promastigotes and amastigotes. Phytomedicine 2009, 16, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Ozer, L.; El-On, J.; Golan-Goldhirsh, A.; Gopas, J. Leishmania major: Anti-leishmanial activity of Nuphar lutea extract mediated by the activation of transcription factor NF-kappaB. Exp. Parasitol. 2010, 126, 510–516. [Google Scholar] [CrossRef]
- Jain, S.; Jacob, M.; Walker, L.; Tekwani, B. Screening North American plant extracts in vitro against Trypanosoma brucei for discovery of new antitrypanosomal drug leads. BMC Complement. Altern. Med. 2016, 16, 131. [Google Scholar] [CrossRef] [Green Version]
- Ozer, J.; Fishman, D.; Eilam, B.; Golan-Goldhirsh, A.; Gopas, J. Anti-Metastatic Effect of Semi-Purified Nuphar Lutea Leaf Extracts. J. Cancer 2017, 8, 1433–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winer, H.; Ozer, J.; Shemer, Y.; Reichenstein, I.; Eilam-Frenkel, B.; Benharroch, D.; Golan-Goldhirsh, A.; Gopas, J. Nuphar lutea Extracts Exhibit Anti-Viral Activity against the Measles Virus. Molecules 2020, 25, 1657. [Google Scholar] [CrossRef] [Green Version]
- Waidha, K.; Anto, N.P.; Jayaram, D.R.; Golan-Goldhirsh, A.; Rajendran, S.; Livneh, E.; Gopas, J. 6,6′-Dihydroxythiobinupharidine (DTBN) Purified from Nuphar lutea Leaves Is an Inhibitor of Protein Kinase C Catalytic Activity. Molecules 2021, 26, 2785. [Google Scholar] [CrossRef]
- De Pasquale, V.; Moles, A.; Pavone, L.M. Cathepsins in the Pathophysiology of Mucopolysaccharidoses: New Perspectives for Therapy. Cells 2020, 9, 979. [Google Scholar] [CrossRef] [Green Version]
- Kopitar-Jerala, N. The role of cysteine proteinases and their inhibitors in the host-pathogen cross talk. Curr. Protein Pept. Sci. 2012, 13, 767–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dana, D.; Pathak, S.K. A Review of Small Molecule Inhibitors and Functional Probes of Human Cathepsin L. Molecules 2020, 25, 698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tada, N.; Jansen, D.J.; Mower, M.P.; Blewett, M.M.; Umotoy, J.C.; Cravatt, B.F.; Wolan, D.W.; Shenvi, R.A. Synthesis and Sulfur Electrophilicity of the Nuphar Thiaspirane Pharmacophore. ACS Cent. Sci. 2016, 2, 401–408. [Google Scholar] [CrossRef]
- Kneller, D.W.; Phillips, G.; O’Neill, H.M.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural plasticity of SARS-CoV-2 3CL M(pro) active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020, 11, 3202. [Google Scholar] [CrossRef]
- Bredenbeek, P.J.; Pachuk, C.J.; Noten, A.F.; Charite, J.; Luytjes, W.; Weiss, S.R.; Spaan, W.J. The primary structure and expression of the second open reading frame of the polymerase gene of the coronavirus MHV-A59; a highly conserved polymerase is expressed by an efficient ribosomal frameshifting mechanism. Nucleic Acids Res. 1990, 18, 1825–1832. [Google Scholar] [CrossRef] [Green Version]
- Thiel, V.; Ivanov, K.A.; Putics, A.; Hertzig, T.; Schelle, B.; Bayer, S.; Weissbrich, B.; Snijder, E.J.; Rabenau, H.; Doerr, H.W.; et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003, 84, 2305–2315. [Google Scholar] [CrossRef] [PubMed]
- Chodera, J.; Lee, A.A.; London, N.; von Delft, F. Crowdsourcing drug discovery for pandemics. Nat. Chem. 2020, 12, 581. [Google Scholar] [CrossRef]
- Lacharity, J.J.; Fournier, J.; Lu, P.; Mailyan, A.K.; Herrmann, A.T.; Zakarian, A. Total Synthesis of Unsymmetrically Oxidized Nuphar Thioalkaloids via Copper-Catalyzed Thiolane Assembly. J. Am. Chem. Soc. 2017, 139, 13272–13275. [Google Scholar] [CrossRef]
- Dalvie, E.D.; Gopas, J.; Golan-Goldhirsh, A.; Osheroff, N. 6,6′-Dihydroxythiobinupharidine as a poison of human type II topoisomerases. Bioorg. Med. Chem. Lett. 2019, 29, 1881–1885. [Google Scholar] [CrossRef]
- Greenspan, P.D.; Clark, K.L.; Tommasi, R.A.; Cowen, S.D.; McQuire, L.W.; Farley, D.L.; van Duzer, J.H.; Goldberg, R.L.; Zhou, H.; Du, Z.; et al. Identification of dipeptidyl nitriles as potent and selective inhibitors of cathepsin B through structure-based drug design. J. Med. Chem. 2001, 44, 4524–4534. [Google Scholar] [CrossRef]
- Giroud, M.; Dietzel, U.; Anselm, L.; Banner, D.; Kuglstatter, A.; Benz, J.; Blanc, J.B.; Gaufreteau, D.; Liu, H.; Lin, X.; et al. Repurposing a Library of Human Cathepsin L Ligands: Identification of Macrocyclic Lactams as Potent Rhodesain and Trypanosoma brucei Inhibitors. J. Med. Chem. 2018, 61, 3350–3369. [Google Scholar] [CrossRef]
- Cai, J.; Fradera, X.; van Zeeland, M.; Dempster, M.; Cameron, K.S.; Bennett, D.J.; Robinson, J.; Popplestone, L.; Baugh, M.; Westwood, P.; et al. 4-(3-Trifluoromethylphenyl)-pyrimidine-2-carbonitrile as cathepsin S inhibitors: N3, not N1 is critically important. Bioorg. Med. Chem. Lett. 2010, 20, 4507–4510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Zhao, Y.; Sun, Y.; Zhang, B.; Wang, H.; Wu, Y.; Zhu, Y.; Zhu, C.; Hu, T.; Du, X.; et al. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 2020, 27, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Arun, K.G.; Sharanya, C.S.; Abhithaj, J.; Francis, D.; Sadasivan, C. Drug repurposing against SARS-CoV-2 using E-pharmacophore based virtual screening, molecular docking and molecular dynamics with main protease as the target. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Abranyi-Balogh, P.; et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020, 11, 5047. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DTBN Binding Energy * ΔG (Kcal/mol) | Cathtepsin Inhibitor’s Binding Energy * ΔG (Kcal/mol) | Cathtepsin Inhibitor’s IC50 Values | H-Bond Interaction of Cathepsins-DTBN (Bond Length Å) | |
|---|---|---|---|---|
| Cathepsin B | −42.57 | −68.3 | 6.8 nM [26] | (i) His111 NH…O of furan ring in DTBN (1.99 Å) (ii) Thr120 C(O)…HO group in DTBN (1.79 Å) (iii) Glu122 (O)C-O…H-O group in DTBN (1.91 Å) |
| Cathepsin L | −42.59 | −51.29 | 3.0 nM [27] | (i) Gln21 C(O)…HO group in DTBN (2.31 Å) (ii) Gln21 NH…OH group in DTBN (1.86 Å) |
| Cathepsin S | −44.3 | −48.4 | 6.0 nM [28] | Val162 C(O)…H-O group in DTBN (1.80 Å) |
| Mpro | −35.87 | −76.14 | 0.67 μM [29] | (i) Glu166 NH…OH group in DTBN (2.13 Å) (ii) Glu166 O-C(O)…HO in DTBN (2.11 Å) |
| Enzyme | iDTBN Covalent Dock Binding Energy * ΔG (Kcal/mol) | Hydrogen Bond Interaction of iDTBN-Cathepsins and Mpro (Bond Length Å) |
|---|---|---|
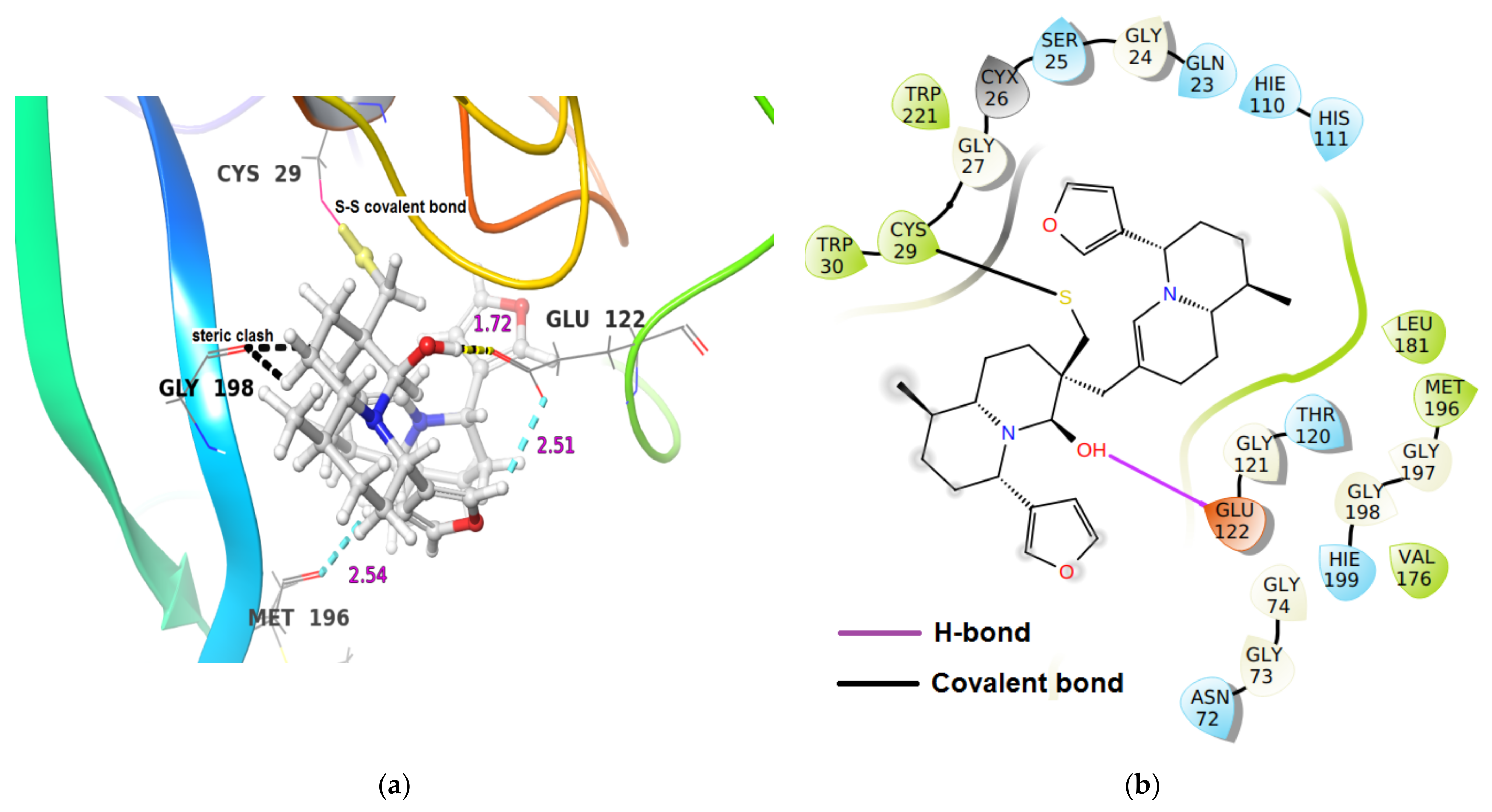
| Cathepsin B | −18.57 | (i) One hydrogen bond interaction Glu122O-C(O)…H-O group proximal to thiaspirane ring of DTBN (1.72 Å). (ii) two C-H…O aromatic hydrogen bond interactions (a) Glu122(O)C-O…H-C of furan ring in DTBN (2.51 Å) and (b) Met196C(O)…H-C of furan ring in DTBN (2.54 Å) |
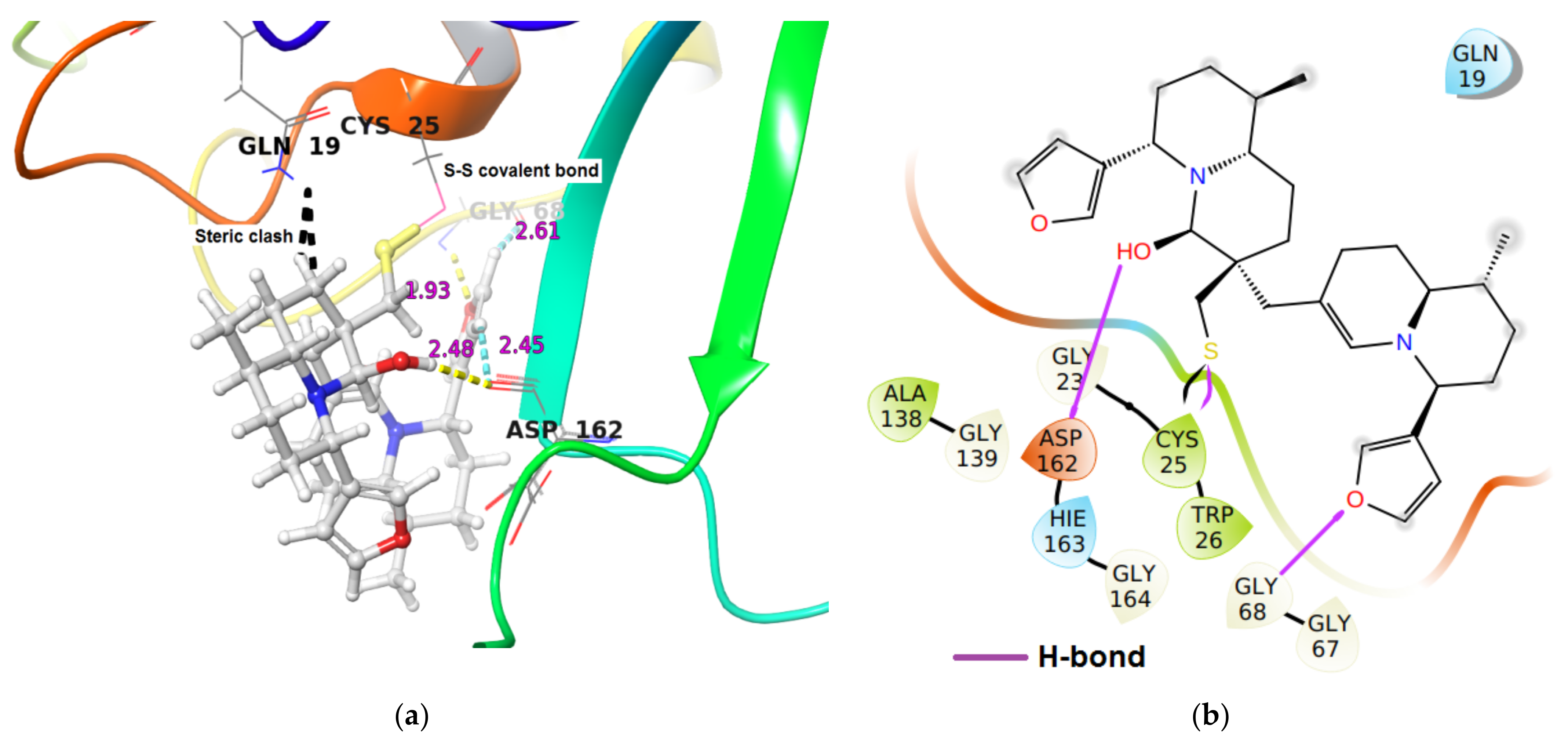
| Cathepsin L | −28.9 | (i) Two hydrogen bond interaction (a) Gly68N-H…O of furan oxygen of DTBN (1.93 Å) (b) Asp162C(O)…H-O of hydroxy group in DTBN (2.48). (ii) two C-H…O aromatic hydrogen bond interactions (a) Gly68C(O)…H-C of furan ring of DTBN (2.61 Å). (b) Asp162C(O)…H-C of furan ring of DTBN (2.45 Å) |
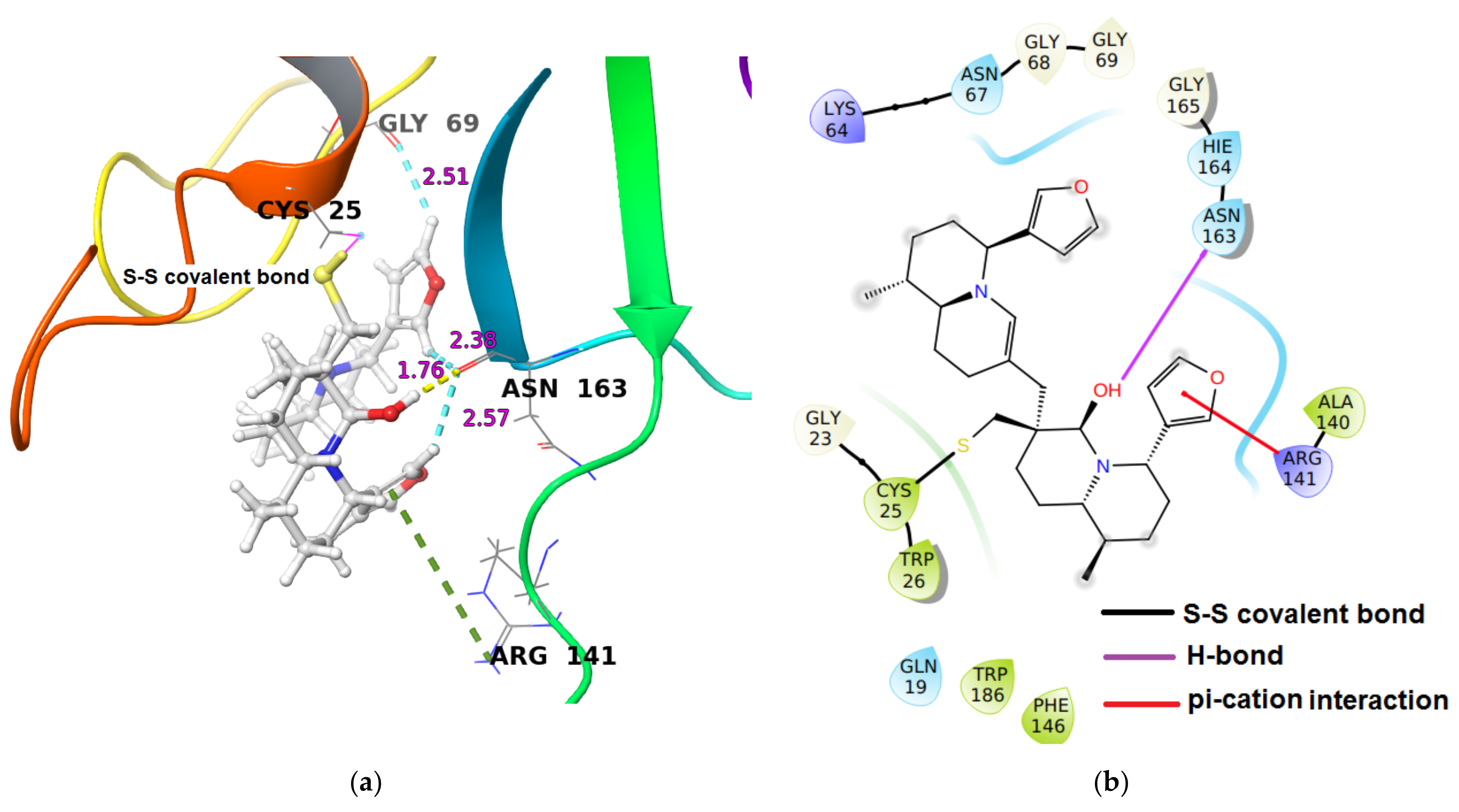
| Cathepsin S | −33.99 | (i) One hydrogen bond interaction: Asn163O-C(O)…H-O of hydroxy group distal from thiaspirane ring in DTBN (1.76 Å), (ii) three C-H…O aromatic hydrogen bond interactions (a) Asn163C(O)…H-C of furan ring of DTBN (2.57 Å). (b) Asn163C(O)…H-C of furan ring of DTBN (2.38 Å). (c) Gly69C(O)…H-C of furan ring of DTBN (2.51 Å). (iii) one π…cation interaction: Arg141H-C=N-H+…π electron of furan ring in DTBN. |
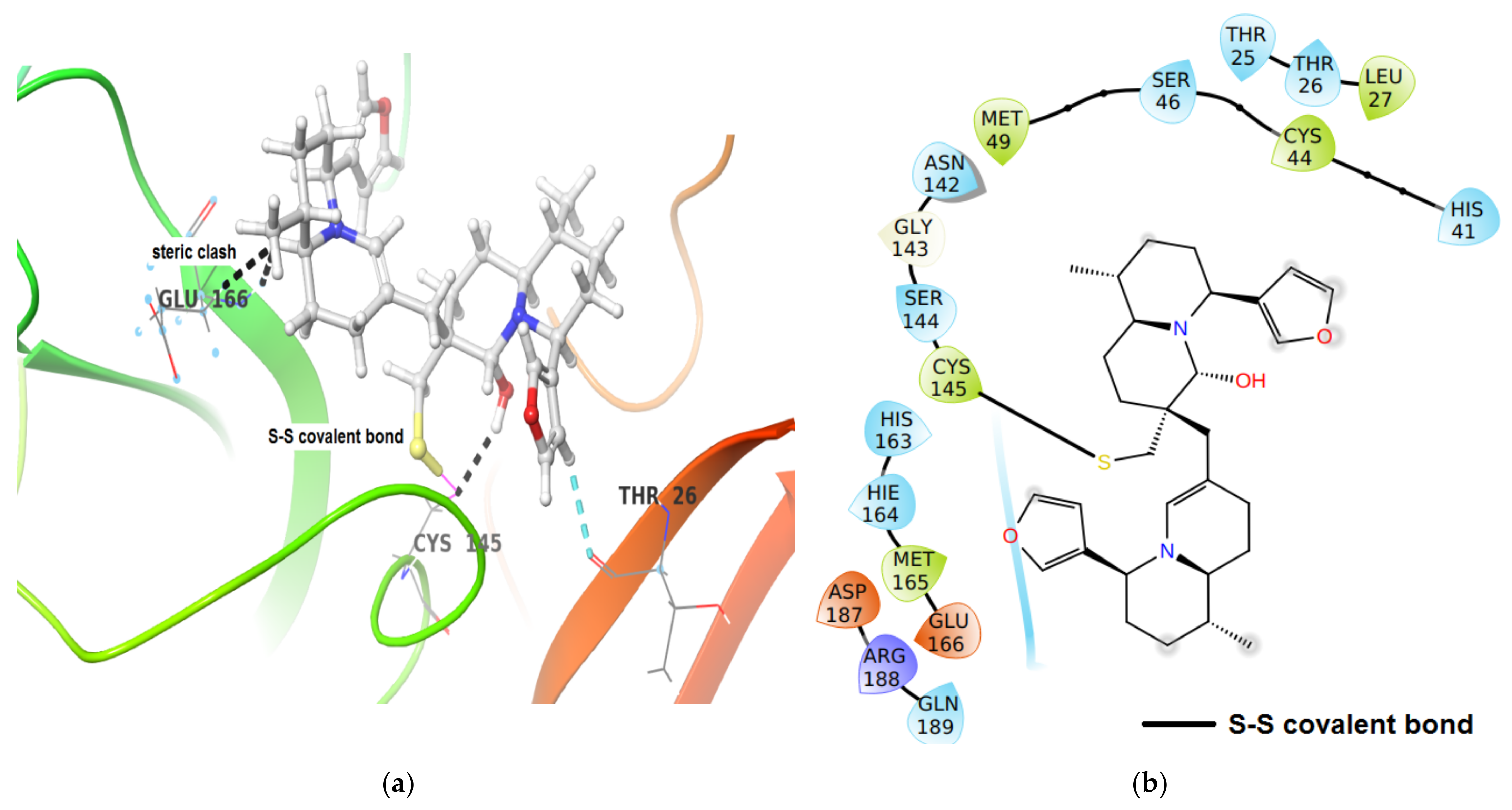
| Mpro | −46.53 | Thr26C(O)…H-C of furan ring of DTBN (2.61 Å). |
| Enzyme | IC50 (µM) (SD) |
|---|---|
| Human Cathepsin S | 3.2 (0.3) |
| Human Cathepsin L | 13.2 (0.6) |
| Human Cathepsin B | 1359.4 (192.0) |
| Papain Latex | 70.4 (0.6) |
| Trypsin * | n.i |
| Calpain * | n.i |
| SARS-CoV-2, Mpro ** | n.i |
| Cathepsin | Active Site Residue Number |
|---|---|
| Cathepsin B | 23, 29, 75, 110, 199, 245 |
| Cathepsin L | 25, 19, 162, 161, 214, 69, 72, 61, 63, 68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waidha, K.; Zurgil, U.; Ben-Zeev, E.; Gopas, J.; Rajendran, S.; Golan-Goldhirsh, A. Inhibition of Cysteine Proteases by 6,6′-Dihydroxythiobinupharidine (DTBN) from Nuphar lutea. Molecules 2021, 26, 4743. https://doi.org/10.3390/molecules26164743
Waidha K, Zurgil U, Ben-Zeev E, Gopas J, Rajendran S, Golan-Goldhirsh A. Inhibition of Cysteine Proteases by 6,6′-Dihydroxythiobinupharidine (DTBN) from Nuphar lutea. Molecules. 2021; 26(16):4743. https://doi.org/10.3390/molecules26164743
Chicago/Turabian StyleWaidha, Kamran, Udi Zurgil, Efrat Ben-Zeev, Jacob Gopas, Saravanakumar Rajendran, and Avi Golan-Goldhirsh. 2021. "Inhibition of Cysteine Proteases by 6,6′-Dihydroxythiobinupharidine (DTBN) from Nuphar lutea" Molecules 26, no. 16: 4743. https://doi.org/10.3390/molecules26164743
APA StyleWaidha, K., Zurgil, U., Ben-Zeev, E., Gopas, J., Rajendran, S., & Golan-Goldhirsh, A. (2021). Inhibition of Cysteine Proteases by 6,6′-Dihydroxythiobinupharidine (DTBN) from Nuphar lutea. Molecules, 26(16), 4743. https://doi.org/10.3390/molecules26164743