
Search for Novel Lead Inhibitors of Yeast Cytochrome bc1, from Drugbank and COCONUT


Abstract
:1. Introduction
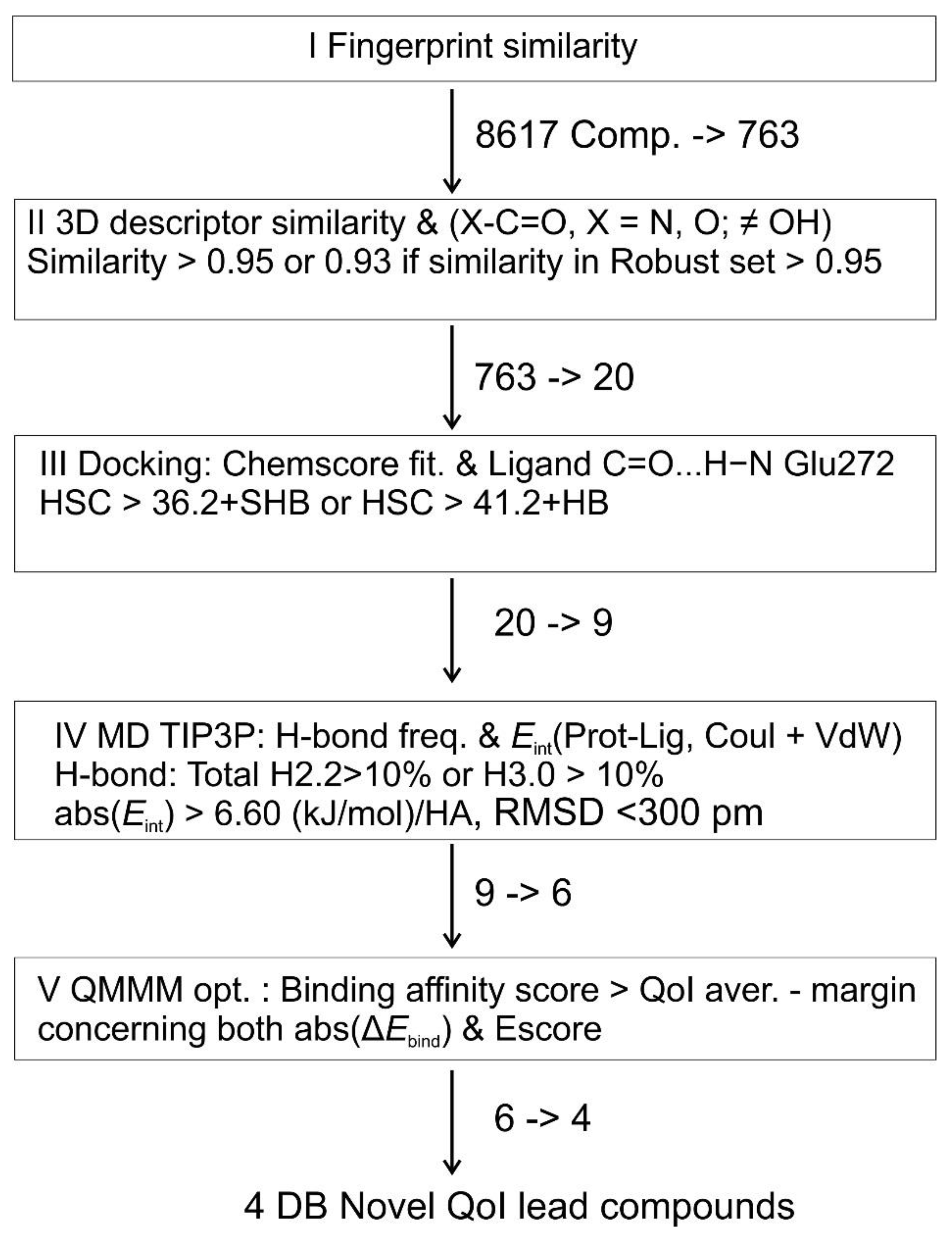
2. Results and Discussion
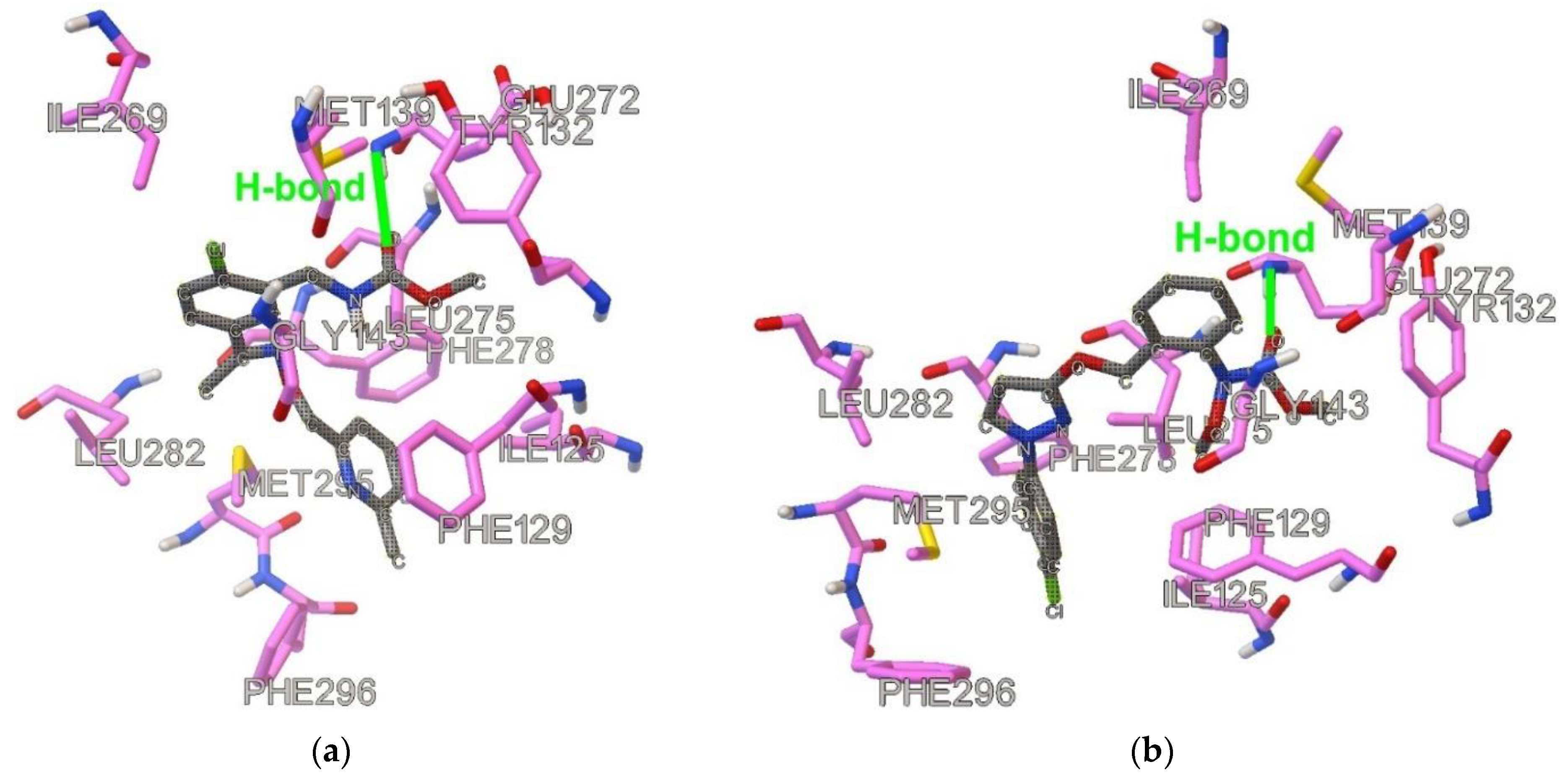
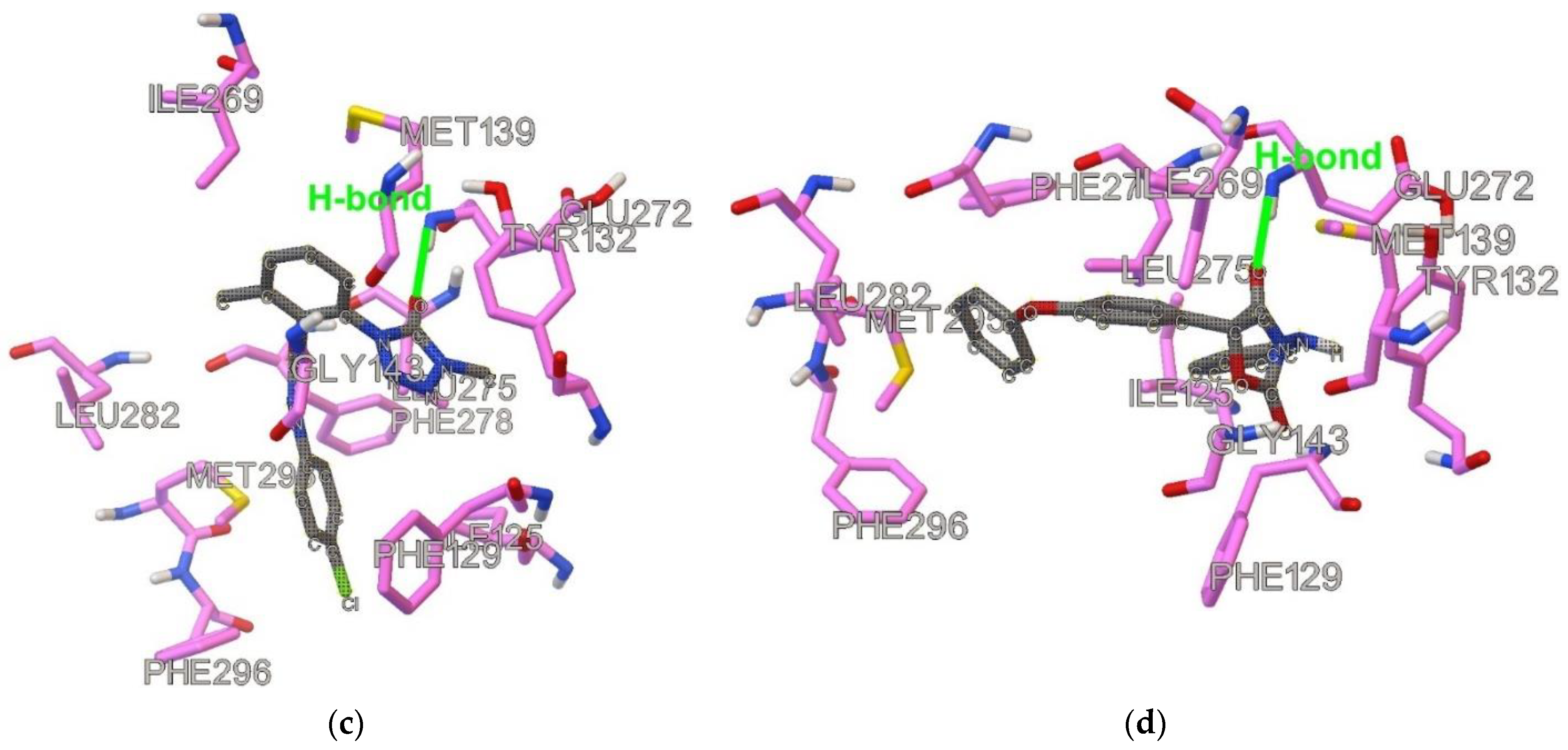
2.1. Similarities and Docking
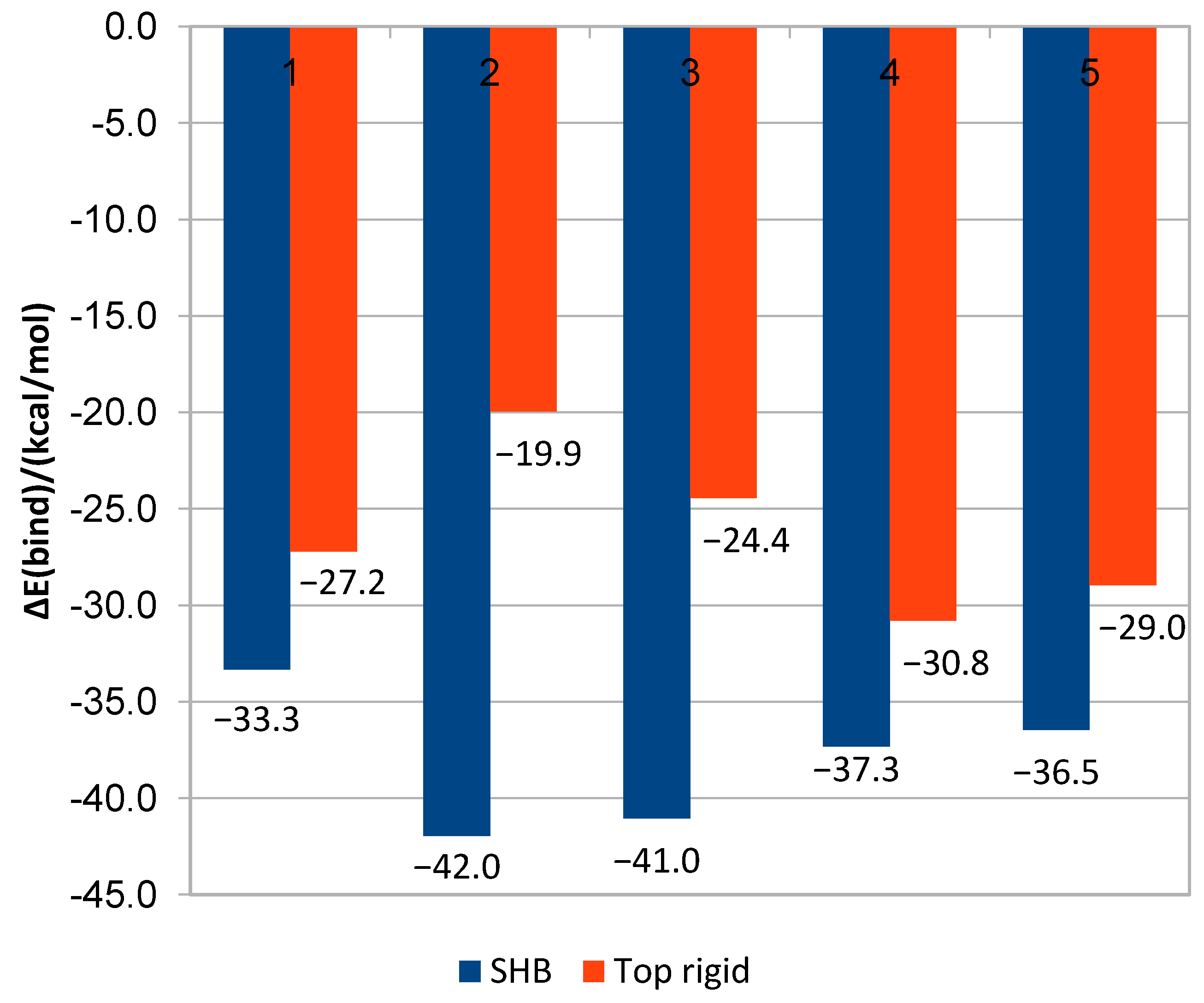
2.2. Results of MD Runs
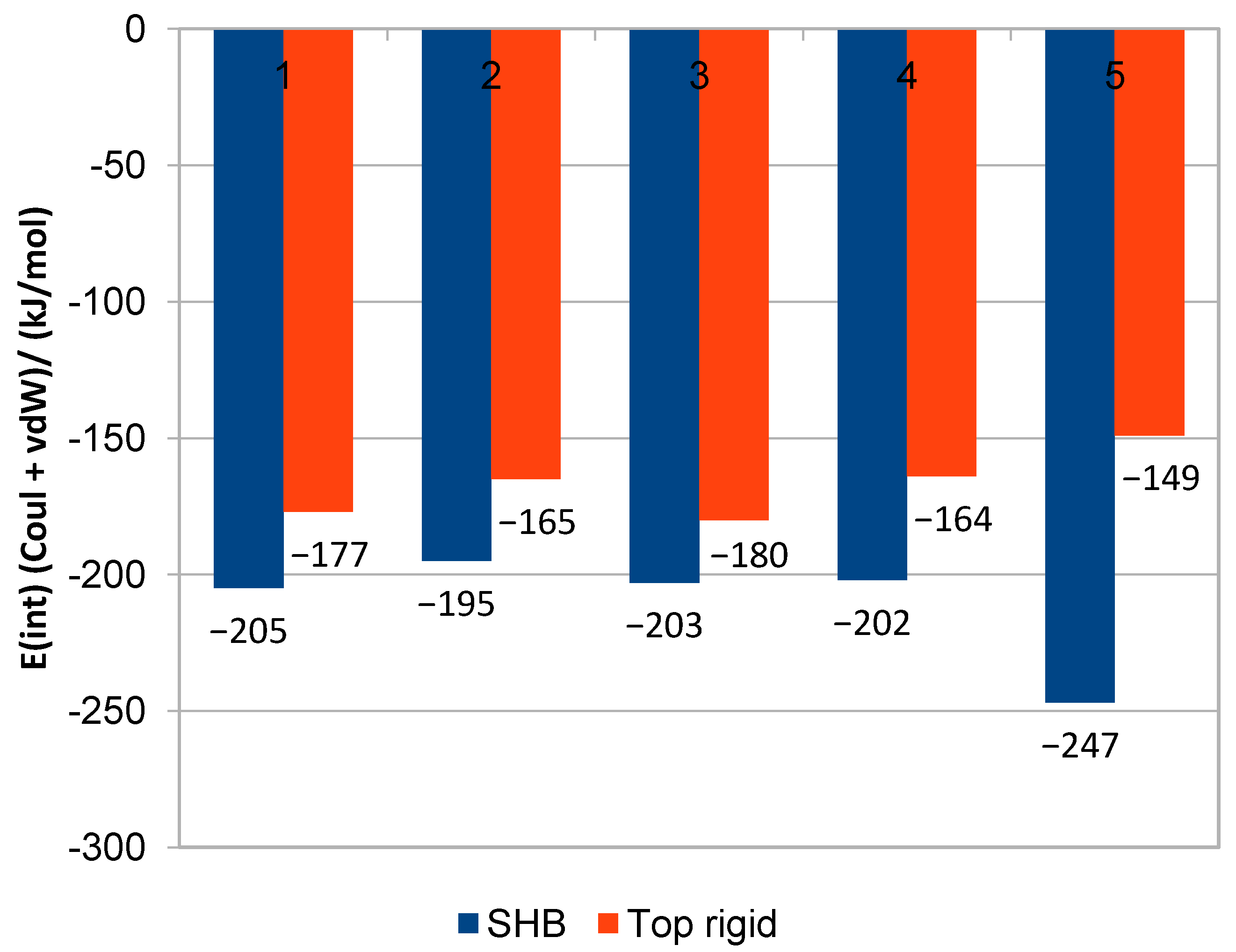
2.3. QM/MM Binding Energy
3. Materials and Methods
3.1. Datasets
3.2. Fingerprint Similarity Filtering
3.3. Pretreatment of the Protein PDB File
3.4. Descriptor Similarity Filtering
3.5. Molecular Docking
3.5.1. Flexible Docking
3.5.2. Rigid Docking
3.6. Molecular Dynamics
3.7. QM/MM Docking Optimization
3.8. Amber MM/PBSA Binding Energy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crofts, A.R. The Cytochrome bc1 Complex: Function in the Context of Structure. Annu. Rev. Physiol. 2004, 66, 689–733. [Google Scholar] [CrossRef] [Green Version]
- FRAC Code List 2020: Fungicides Sorted by Mode of Action. Available online: http://www.frac.info/ (accessed on 25 September 2020).
- Gao, X.; Wen, X.; Yu, C.; Esser, L.; Tsao, S.; Quinn, B.; Zhang, L.; Yu, L.; Xia, D. The Crystal Structure of Mitochondrial Cytochrome bc1 in Complex with Famoxadone: The Role of Aromatic-Aromatic Interaction in Inhibition. Biochemistry 2002, 41, 11692–11702. [Google Scholar] [CrossRef]
- Hao, G.-F.; Wang, F.; Li, H.; Zhu, X.-L.; Yang, W.-C.; Huang, L.-S.; Wu, J.-W.; Berry, E.A.; Yang, G.-F. Computational Discovery of Picomolar Qo Site Inhibitors of Cytochrome bc1 Complex. J. Am. Chem. Soc. 2012, 134, 11168–11176. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, S.; Kong, X.; Cao, L.; Tian, S.; Ye, Y.; Qiao, C. Design, synthesis and fungicidal evaluation of novel pyraclostrobin analogues. Bioorg. Med. Chem. 2018, 26, 875–883. [Google Scholar]
- Fisher, N.; Meunier, B. Re-examination of inhibitor resistance conferred by Qo-site mutations in cytochrome b using yeast as a model system. Pest Manag. Sci. 2005, 61, 973–978. [Google Scholar] [CrossRef]
- Matsuzaki, Y.; Yoshimoto, Y.; Arimori, S.; Kiguchi, S.; Harada, T.; Iwahashi, F. Discovery of metyltetraprole: Identification of tetrazolinone pharmacophore to overcome QoI resistance. Bioorg. Med. Chem. 2020, 28, 115211. [Google Scholar] [CrossRef]
- Cai, N.; He, L.; Wang, K.; Feng, Z.; Cui, Z.; Ji, M.; Qi, Z.; Qin, P.; Li, X. Novel sulfonamides against Botrytis cinerea with no positive cross-resistance to commercial fungicides: Design, synthesis and SAR study. Bioorg. Med. Chem. Lett. 2020, 30, 126859. [Google Scholar] [CrossRef]
- Sorokina, M.; Merseburger, P.; Rajan, K.; Yirik, M.A.; Steinbeck, C. COCONUT online: Collection of Open Natural Products database. J. Cheminform. 2021, 13, 2. [Google Scholar] [CrossRef]
- Musso, L.; Fabbrini, A.; Dallavalle, S. Natural Compound-Derived Cytochrome bc1 Complex Inhibitors as Antifungal Agents. Molecules 2020, 25, 4582. [Google Scholar] [CrossRef]
- Zheng, Y.-J. Molecular basis for the enantioselective binding of a novel class of cytochrome bc1 complex inhibitors. J. Mol. Graph. Model. 2006, 25, 71–76. [Google Scholar] [CrossRef]
- Estève, K.; Poupot, C.; Dabert, P.; Mietton-Peuchot, M.; Milisic, V. A Saccharomyces cerevisiae-based bioassay for assessing pesticide toxicity. J. Ind. Microbiol. Biotechnol. 2009, 36, 1529–1534. [Google Scholar] [CrossRef]
- González-Rodríguez, R.M.; González-Barreiro, C.; Rial-Otero, R.; Regueiro, J.; Torrado-Agrasar, A.; Martínez-Carballo, E.; Cancho-Grande, B. Influence of new fungicides—Metiram and pyraclostrobin—On Saccharomyces cerevisiae yeast growth and alcoholic fermentation course for wine production. CYTA J. Food. 2011, 9, 329–334. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.-L.; Wang, L.; Zhu, X.-L.; Huang, X.; Zhan, C.-G.; Wu, J.-W.; Yang, G.-F. Subnanomolar Inhibitor of Cytochrome bc1 Complex Designed by Optimizing Interaction with Conformationally Flexible Residues. J. Am. Chem. Soc. 2010, 132, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakawa, A.; Kasai, Y.; Yamazaki, K.; Iwahashi, F. Features of interactions responsible for antifungal activity against resistant type cytochrome bc1: A data-driven analysis based on the binding free energy at the atomic level. PLoS ONE 2018, 13, e0207673. [Google Scholar] [CrossRef] [PubMed]
- GROMACS Tutorial. Available online: http://www.mdtutorials.com/gmx/complex/09_analysis.html (accessed on 12 December 2020).
- Suemoto, H.; Matsuzaki, Y.; Iwahashi, F. Metyltetraprole, a novel putative complex III inhibitor, targets known QoI-resistant strains of Zymoseptoria tritici and Pyrenophora teres. Pest Manag. Sci. 2019, 75, 1181–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- COCONUT, Collection of Open Natural Products. Available online: https://coconut.naturalproducts.net/compound/coconut_id/CNP0061106 (accessed on 2 February 2021).
- Genheden, S.; Rede, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Lancaster, C.R.; Hunte, C.; Kelley, J.; Trumpower, B.L.; Ditchfield, R. A Comparison of Stigmatellin Conformations, Free and Bound to the Photosynthetic Reaction Center and the Cytochrome bc(1) Complex. J. Mol. Biol. 2007, 368, 197–208. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guterres, H.; Im, W. Improving Protein-Ligand Docking Results with High-Throughput Molecular Dynamics Simulations. J. Chem. Inf. Model. 2020, 60, 2189–2198. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 8, e1327. [Google Scholar] [CrossRef]
- DFT ORCA manual, version 4.2.0., ORCA MM Module (chapter 8.18) and ORCA QM/MM Implementation (chapter 8.19) with detailed instructions how to prepare and run your QM/MM system pages 319–341.
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, B.; Gerber, P.; Schulz-Gasch, T.; Stahl, M. Validation and Use of the MM-PBSA Approach for Drug Discovery. J. Med. Chem. 2005, 48, 4040–4048. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulation. Nucleic Acids Res. 2012, 40, W537–W541. Available online: http://biophysics.cs.vt.edu/ (accessed on 3 March 2020). [CrossRef] [Green Version]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; et al. AMBER 2021, University of California, San Francisco.
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3312–3321. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
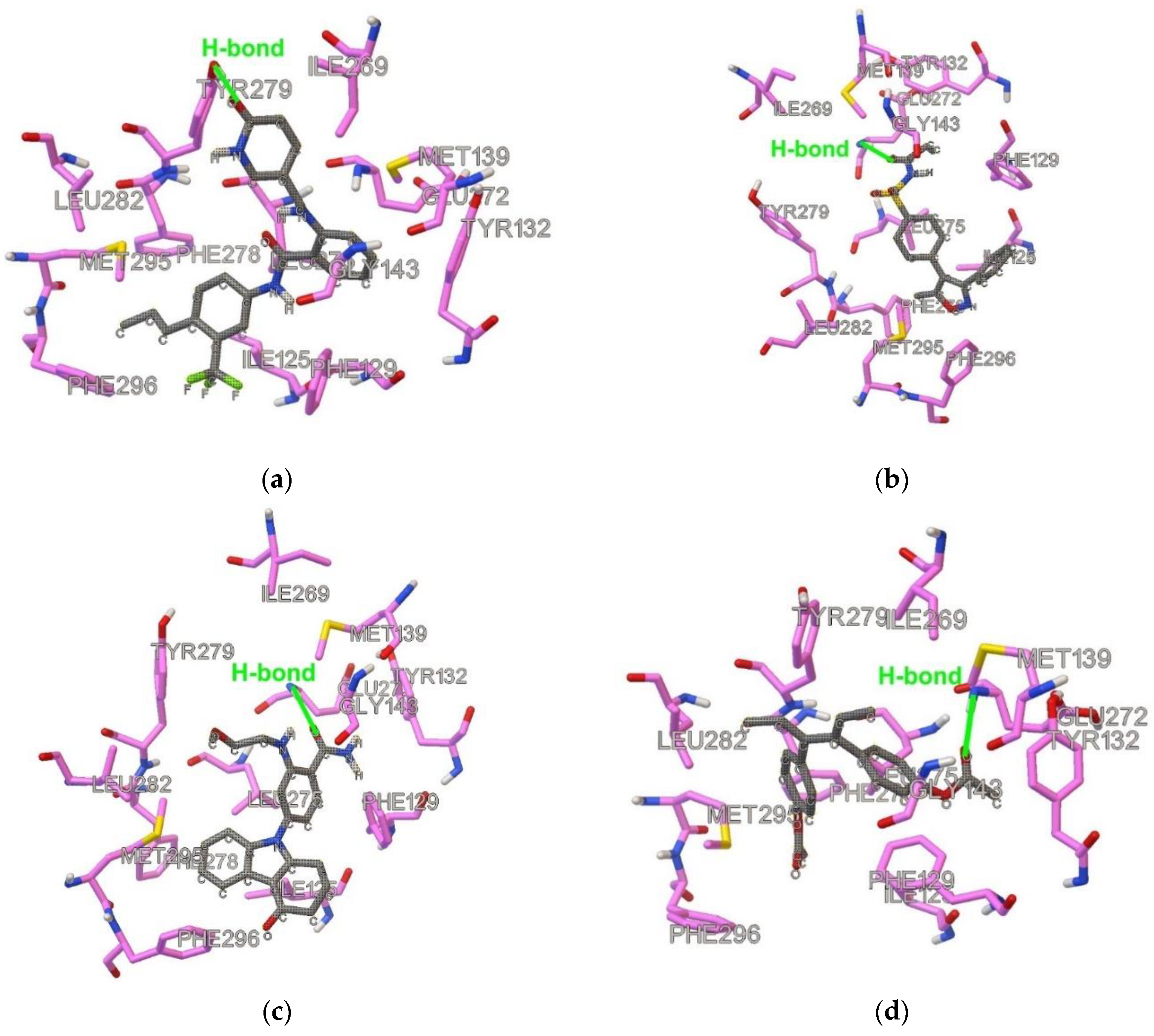{kind=link}
{kind=link}
{kind=link}
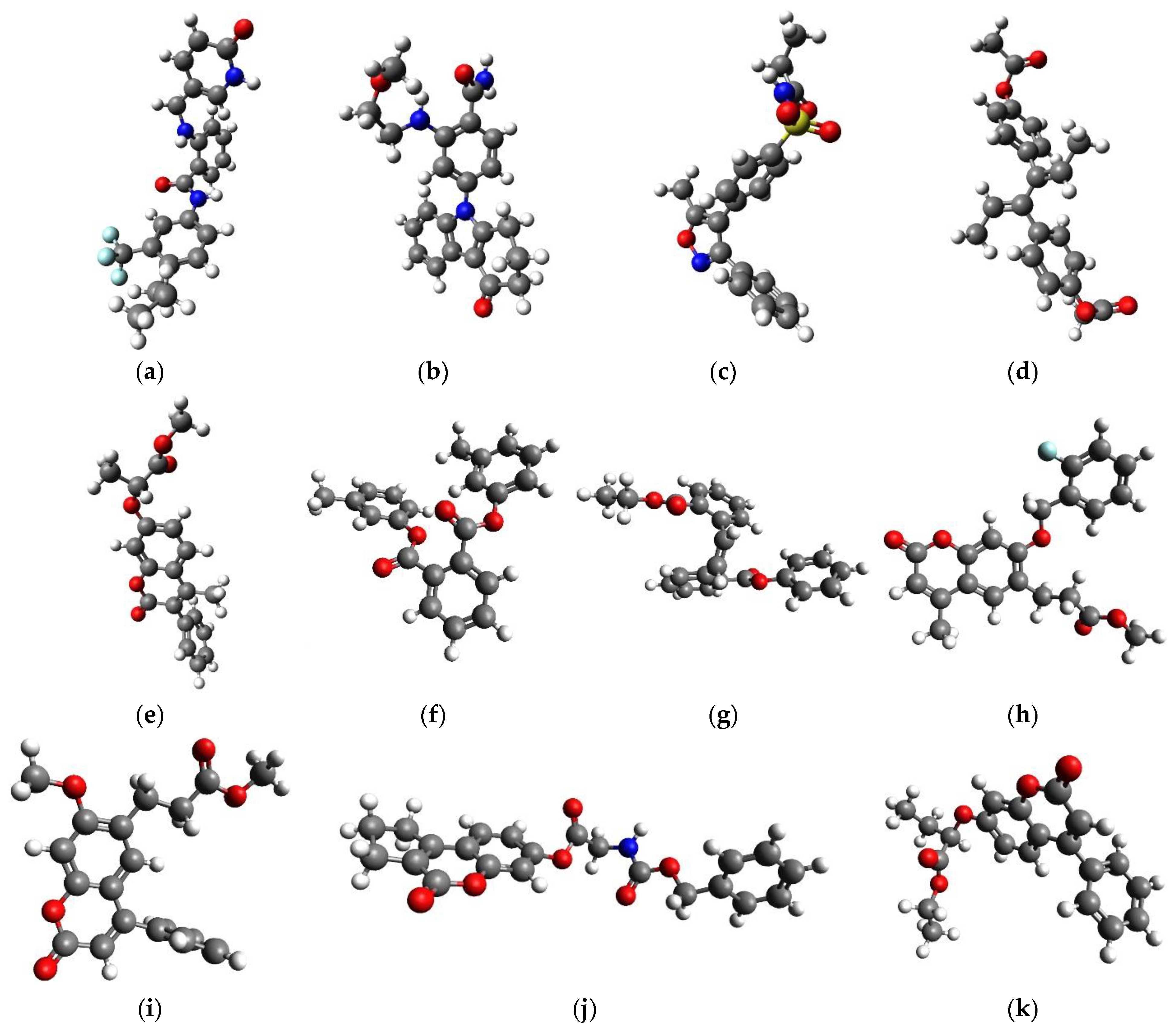{kind=link}
| Compound | HSC | SHB | Compound | HSC | SHB | Compound | HSC | SHB |
|---|---|---|---|---|---|---|---|---|
| azoxystrobin | 32.3 | 30.4 | picoxystrobin | 30.4 | 27.3 | famoxadone | 40.8 | 40.8 (a) |
| metominostrobin | 31.7 | 31.7 (a) | pyrametostrobin | 34.9 | 34.9 (a) | coumoxystrobin | 42.7 | 35.4 |
| pyraclostrobin | 35.4 | 35.3 | pyribencarb | 39.0 | 37.5 | enoxastrobin | 40.8 | 34.2 |
| fenamidone | 33.7 | - (c) | triclopyricarb | 29.5 | 29.5 (a) | mandestrobin | 36.7 | 36.7 (a) |
| kresoxim-methyl | 33.7 | 32.5 | fluoxastrobin | 32.0 | - | dimoxystrobin | 37.8 | 37.8 (a) |
| trifloxystrobin | 35.8 | 35.8 (a) | pyraoxystrobin | 39.9 | 34.8 | flufenoxystrobin | 36.3 | 36.3t (a,b) |
| orysastrobin | 30.9 | 27.3 | fenaminstrobin | 41.2 (a) | 35.6 | metyltetraprole | 37.7 | 37.7 (a) |
| stigmatellin | 46.1 | - | fungicide average | 36.2 (a) | ||||
| DB Hits (0.95 similarity): | ||||||||
| DB00682 | 31.8 | - | DB07831 | 42.3 | 37.8 | DB09199 | 42.0 | (d) |
| DB00946 | 33.2 | - | DB07943 | 37.4 | 34.2 | DB12398 | 33.3 | - |
| DB01418 | 33.9 | - | DB08242 | 38.4 | 35.2 | DB13657 | 30.9 | - |
| DB02660 | 31.3 | - | DB08268 | 33.9 | - | DB14055 | 31.9 | - |
| DB04930 | 43.3 | - (d) | DB08315 | 33.7 | - | DB14668 | 39.5 | 37.5 |
| DB07132 | 32.3 | - | DB08439 | 36.9 | 34.8 | DB07181 | 44.1 | 42.9 |
| DB07704 | 33.6 | - | DB08557 | 39.8 | 39.8t | |||
| Compound | Total H2.2 | H2.2 Glu272 | H3.0 Glu272 | Compound | Total H2.2 | H2.2 Glu272 | H3.0 Glu272 |
|---|---|---|---|---|---|---|---|
| azoxystrobin | 43.68 | 43.68 | 88.52 | dimoxystrobin | 60.82 | 60.38 | 99.74 |
| metominostrobin | 34.61 | 33.48 | 73.98 | flufenoxystrobin | 84.32 | 84.32 | 99.53 |
| Pyraclostrobin | 16.68 | 16.68 | 58.65 | fenaminstrobin | 49.95 | 49.95 | 92.27 |
| kresoxim-methyl | 76.38 | 76.38 | 99.34 | ||||
| trifloxystrobin | 85.03 | 85.03 | 100 | DB04930 | 0 | 0 | 0.02 |
| orysastrobin | 10.63 | 10.63 | 15.15 | DB07831 | 5.72 | 5.71 | 7.41 |
| picoxystrobin | 2.28 | 2.28 | 15.20 | DB07831 (c) | 3.98 | 3.91 | 5.20 |
| pyrametostrobin | 5.74 | 5.74 | 69.54 | DB07831 * | 15.48 | 0 | 0 |
| pyribencarb | 63.32 | 63.21 | 99.38 | DB07943 | 25.01 | 23.65 | 82.50 |
| triclopyricarb | 0.29 | 0.29 | 12.71 | DB08439 (C=O) | 59.50 | 59.50 | 97.79 |
| Stigmatellin (a) | 13.43 | 0.01 | 0.50 | DB08439 (S=O) | 39.89 | 39.89 | 100 |
| famoxadone | 35.78 | 33.98 | 97.60 | DB08557 | 36.70 | 3.85 | 14.72 |
| pyraoxystrobin | 13.11 | 13.11 | 79.09 | DB14668 | 43.28 | 43.28 | 67.38 |
| Coumoxystrobin (b) | 30.55 | 0.27 | 1.04 | DB07181 | 100 | 64.83 | 99.84 |
| enoxastrobin | 81.94 | 81.94 | 99.95 | DB08242 | 2.54 | 2.12 | 21.70 |
| mandestrobin | 52.46 | 52.29 | 98.23 | DB09199 | 2.34 | 0 | 0 |
| metyltetraprole | 95.10 | 95.10 | 100 |
| Compound | HAC | E(int)/kJ/mol | E(int)/HAC | Compound | HAC | E(int)/kJ/mol | E(int)/HAC |
|---|---|---|---|---|---|---|---|
| azoxystrobin | 30 | −228 ± 18 | −7.60 ± 0.60 | enoxastrobin | 28 | −247 ± 16 | −8.82 ± 0.57 |
| metominostrobin | 21 | −156 ± 15 | −7.43 ± 0.71 | Enoxastrobin (b) | 28 | −149 ± 15 | - |
| fenaminstrobin | 29 | −227 ± 14 | −7.83 ± 0.48 | mandestrobin | 23 | −187 ± 12 | −8.13 ± 0.52 |
| Pyraclostrobin | 27 | −195 ± 15 | −7.22 ± 0.56 | metyltetraprole | 28 | −233 ± 12 | −8.32 ± 0.43 |
| Pyraclostrobin (b) | 27 | −167 ± 12 | - | dimoxystrobin | 24 | −226 ± 13 | −9.42 ± 0.54 |
| kresoxim-methyl | 23 | −206 ± 12 | −8.96 ± 0.52 | flufenoxystrobin | 27 | −221 ± 14 | −8.19 ± 0.54 |
| trifloxystrobin | 29 | −232 ± 14 | −8.00 ± 0.48 | ||||
| orysastrobin | 28 | −192 ± 13 | −6.86 ± 0.46 | DB04930 | 26 | −173 ± 10 | −6.65 ± 0.38 |
| picoxystrobin | 26 | −183 ± 13 | −7.03 ± 0.50 | DB07831 | 31 | −169 ± 13 | - |
| pyrametostrobin | 28 | −205 ± 13 | −7.32 ± 0.46 | DB07831 (c) | 31 | −175 ± 13 | - |
| Pyrametostrobin (b) | 28 | −177 ± 13 | - | DB07831 (a) | 31 | −211 ± 13 | −6.81 ± 0.42 |
| pyribencarb | 25 | −203 ± 12 | −8.12 ± 0.48 | DB07943 | 28 | −198 ± 13 | −7.07 ± 0.46 |
| Pyribencarb (b) | 25 | −180 ± 13 | - | DB08439 (C=O) | 26 | −188 ±11 | −7.23 ± 0.42 |
| triclopyricarb | 24 | −156 ± 11 | −6.50 ± 0.46 | DB08439 (S=O) | 26 | −174 ± 11 | - |
| Stigmatellin (b) | 37 | −219 ± 17 | −5.92 ± 0.46 | DB08557 | 28 | −185 ± 13 | −6.61 ± 0.46 |
| famoxadone | 28 | −202 ± 11 | −7.21 ± 0.39 | DB14668 | 26 | −172 ± 16 | −6.62 ± 0.62 |
| Famoxadone (b) | 28 | −164 ± 10 | - | DB07181 | 26 | −191 ± 14 | −7.35 ± 0.54 |
| pyraoxystrobin | 29 | −219 ± 15 | −7.55 ± 0.52 | DB08242 | 27 | −175 ± 12 | −6.48 ± 0.44 |
| coumoxystrobin | 32 | −172 ± 22 | −5.38 ± 0.69 | DB09199 | 27 | −152 ± 11 | −5.63 ± 0.41 |
| B97-D3/def2-SVP//B97-D3/def2-SVP | B97-D3/def2-SVP//B97-D3/3-21G | |||||||
|---|---|---|---|---|---|---|---|---|
| Compound | HAC | ΔE Bind | Escore ΔE/HAC | ΔG | HAC | ΔE bind | Escore ΔE/HAC | ΔG |
| azoxystrobin SHB | 30 | −26.4 | −0.88 | −11.1 | 30 | −24.3 | −0.81 | −1.0 |
| azoxystrobin HSC | 30 | −32.4 | −1.08 | −17.1 | 30 | −29.8 | −0.99 | −6.5 |
| metyltetraprole | 28 | −48.2 | −1.72 | −34.7 | 28 | −40.3 | −1.44 | −18.2 |
| pyraclostrobin | 27 | −40.5 | −1.50 | −36.2 | 27 | −42.0 | −1.55 | −29.0 |
| trifloxystrobin | 29 | −29.7 | −1.02 | −6.8 | 29 | −22.7 | −0.78 | +8.9 |
| pyribencarb | 25 | −44.0 | −1.76 | −29.4 | 25 | −41.0 | −1.64 | −19.5 |
| fenaminstrobin | 29 | −29.0 | −1.00 | −3.1 | 29 | −26.8 | −0.92 | −0.9 |
| famoxadone | 28 | −54.1 | −1.93 | −57.1 | 28 | −37.3 | −1.33 | −36.6 |
| enoxastrobin | 28 | −36.9 | −1.32 | −19.2 | 28 | −36.5 | −1.30 | −10.0 |
| mandestrobin | 23 | −22.7 | −0.99 | −8.7 | 23 | - | - | - |
| dimoxystrobin | 24 | −56.2 | −2.34 | −34.0 | 24 | −49.6 | −2.07 | −21.4 |
| flufenoxystrobin | 27 | −43.8 | −1.62 | −38.1 | 27 | −36.8 | −1.36 | −22.4 |
| kresoxim-methyl | 23 | −27.1 | −1.18 | - | 23 | −23.1 | −1.00 | - |
| Stigmatellin*(do) | 37 | −59.4 | −1.61 | −25.9 | 37 | −48.9 | −1.26 | −12.2 |
| Stigmatellin*(cr) | 37 | −66.6 | −1.80 | −37.1 | 37 | −46.5 | −1.32 | −9.8 |
| Average for QoI | - | −41.1 | −1.45 | −25.5 | - | −36.1 | −1.27 | −13.7 |
| Error margin (a) | - | 6.0 | 0.2 | >6 | - | 5.5 | 0.18 | >6 |
| Hits: | ||||||||
| DB07831 * | 31 | −57.1 | −1.84 | −56.8 | 31 | −59.5 | −1.92 | −50.6 |
| DB07943 | 28 | −31.0 | −1.11 | −16.7 | 28 | −23.8 | −0.85 | −0.8 |
| DB08439 * | 26 | −41.7 | −1.60 | −22.3 | 26 | −36.3 | −1.40 | −8.3 |
| DB08557 * | 28 | −38.7 | −1.38 | −25.2 | 28 | −36.8 | −1.31 | −23.2 |
| DB07181 | 26 | −23.1 | −0.89 | −15.1 | 26 | −17.9 | −0.69 | −1.2 |
| DB14668 * | 26 | −54.9 | −2.11 | −30.8 | 26 | −41.2 | −1.58 | −8.4 |
| CNP0061106 * | 25 | −37.3 | −1.49 | −39.1 | 25 | −34.1 | −1.36 | −27.2 |
| CNP0083976 * | 26 | −55.2 | −2.12 | −53.2 | 26 | −56.1 | −2.16 | −45.4 |
| CNP0105092 * | 28 | −37.0 | −1.32 | −20.9 | 28 | −34.4 | −1.23 | −9.6 |
| CNP0153569 * | 27 | −42.2 | −1.56 | −29.3 | 27 | −36.8 | −1.36 | −15.3 |
| CNP0330896 * | 25 | −39.9 | −1.59 | −15.7 | 25 | −38.0 | −1.52 | −5.2 |
| CNP0361420 * | 29 | −43.0 | −1.48 | −36.7 | 29 | −45.3 | −1.56 | −30.4 |
| CNP0364473 * | 26 | −53.4 | −2.05 | −45.6 | 26 | −43.0 | −1.65 | −26.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jović, O.; Šmuc, T. Search for Novel Lead Inhibitors of Yeast Cytochrome bc1, from Drugbank and COCONUT. Molecules 2021, 26, 4323. https://doi.org/10.3390/molecules26144323
Jović O, Šmuc T. Search for Novel Lead Inhibitors of Yeast Cytochrome bc1, from Drugbank and COCONUT. Molecules. 2021; 26(14):4323. https://doi.org/10.3390/molecules26144323
Chicago/Turabian StyleJović, Ozren, and Tomislav Šmuc. 2021. "Search for Novel Lead Inhibitors of Yeast Cytochrome bc1, from Drugbank and COCONUT" Molecules 26, no. 14: 4323. https://doi.org/10.3390/molecules26144323
APA StyleJović, O., & Šmuc, T. (2021). Search for Novel Lead Inhibitors of Yeast Cytochrome bc1, from Drugbank and COCONUT. Molecules, 26(14), 4323. https://doi.org/10.3390/molecules26144323