
Targeting 3CLpro and SARS-CoV-2 RdRp by Amphimedon sp. Metabolites: A Computational Study

 , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Sponge Material
2.2. Extraction and Isolation
2.3. Sequence Alignment and Modeling
2.4. Molecular Docking Methodology
2.5. Prediction of Pharmacokinetic Properties and Toxicological Properties ADME/T
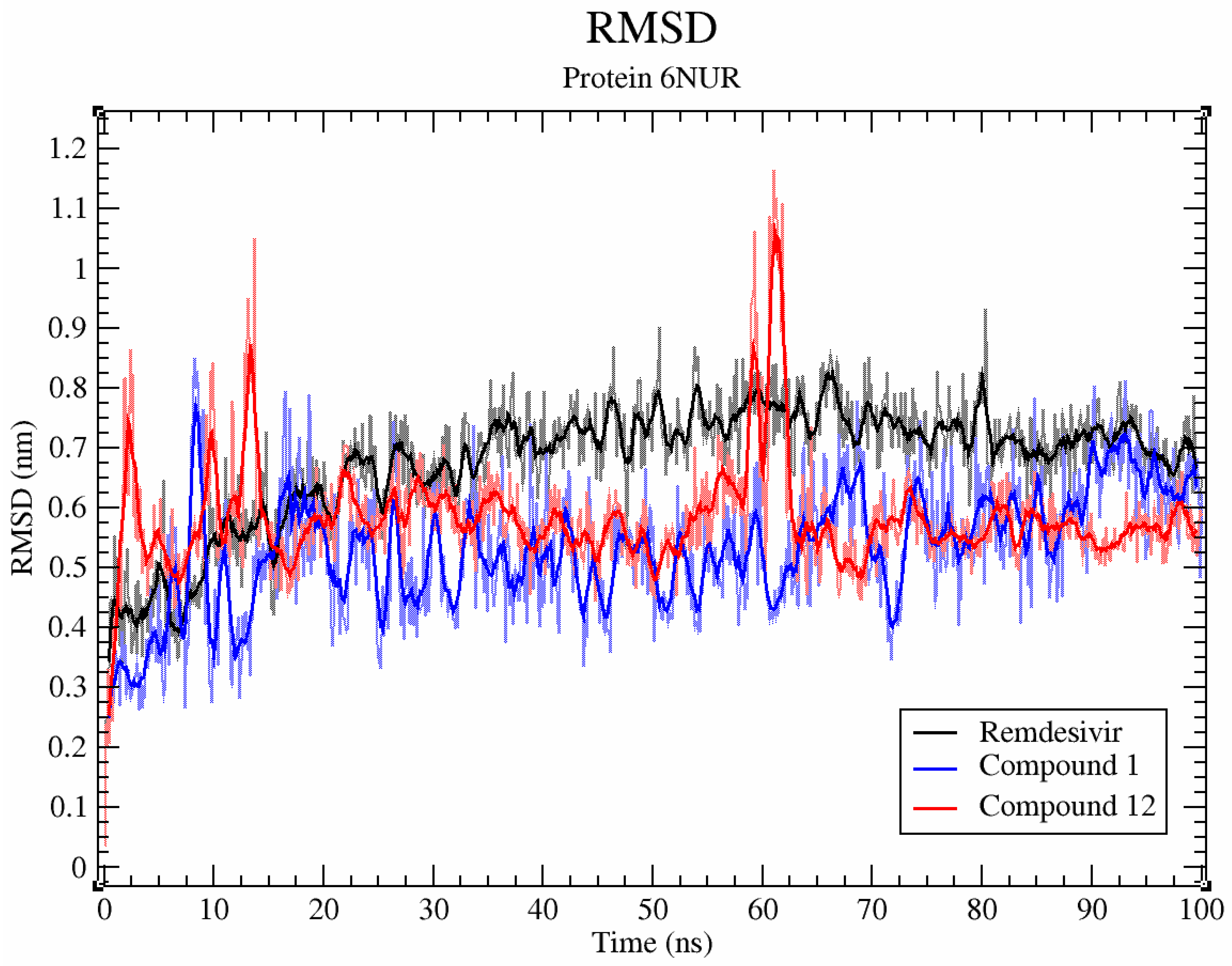
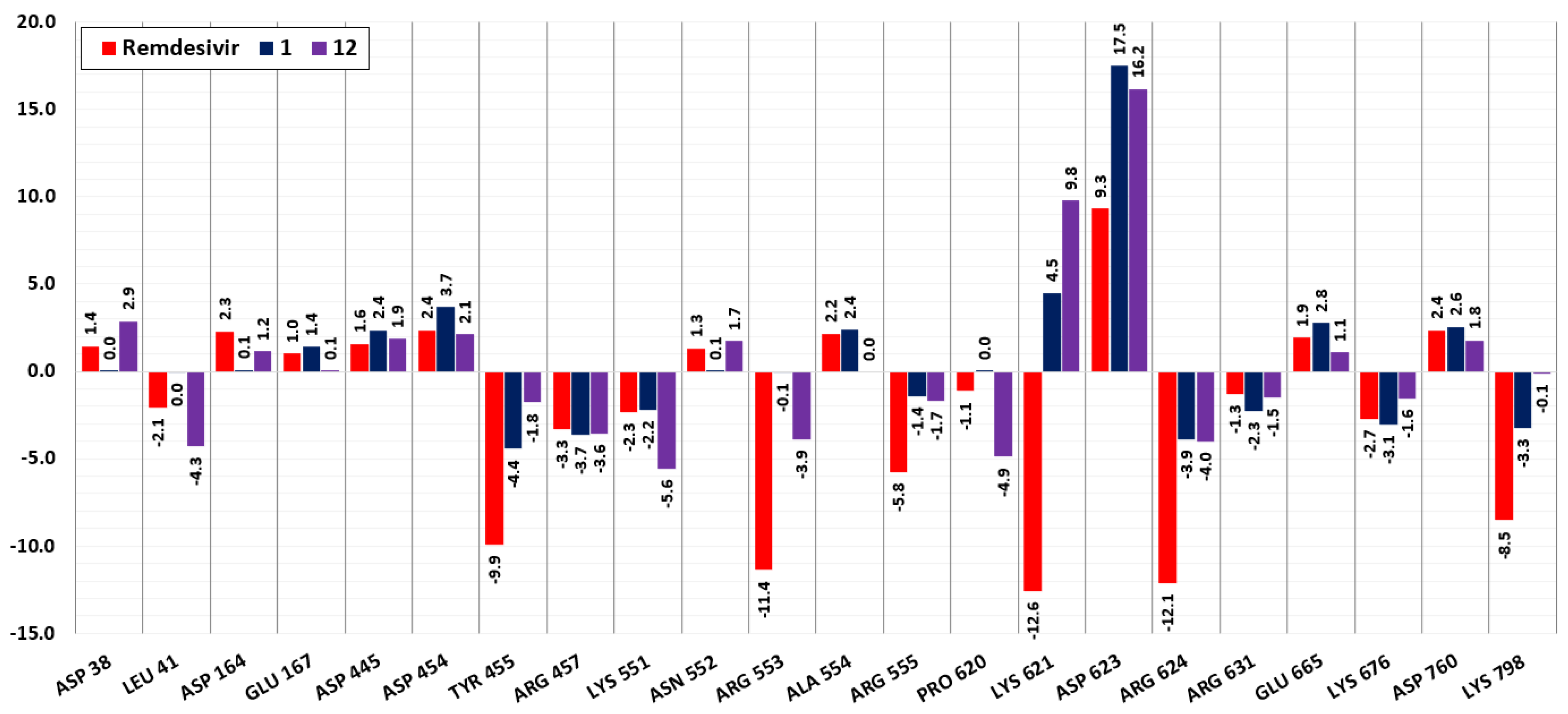
2.6. Molecular Dynamics Simulations
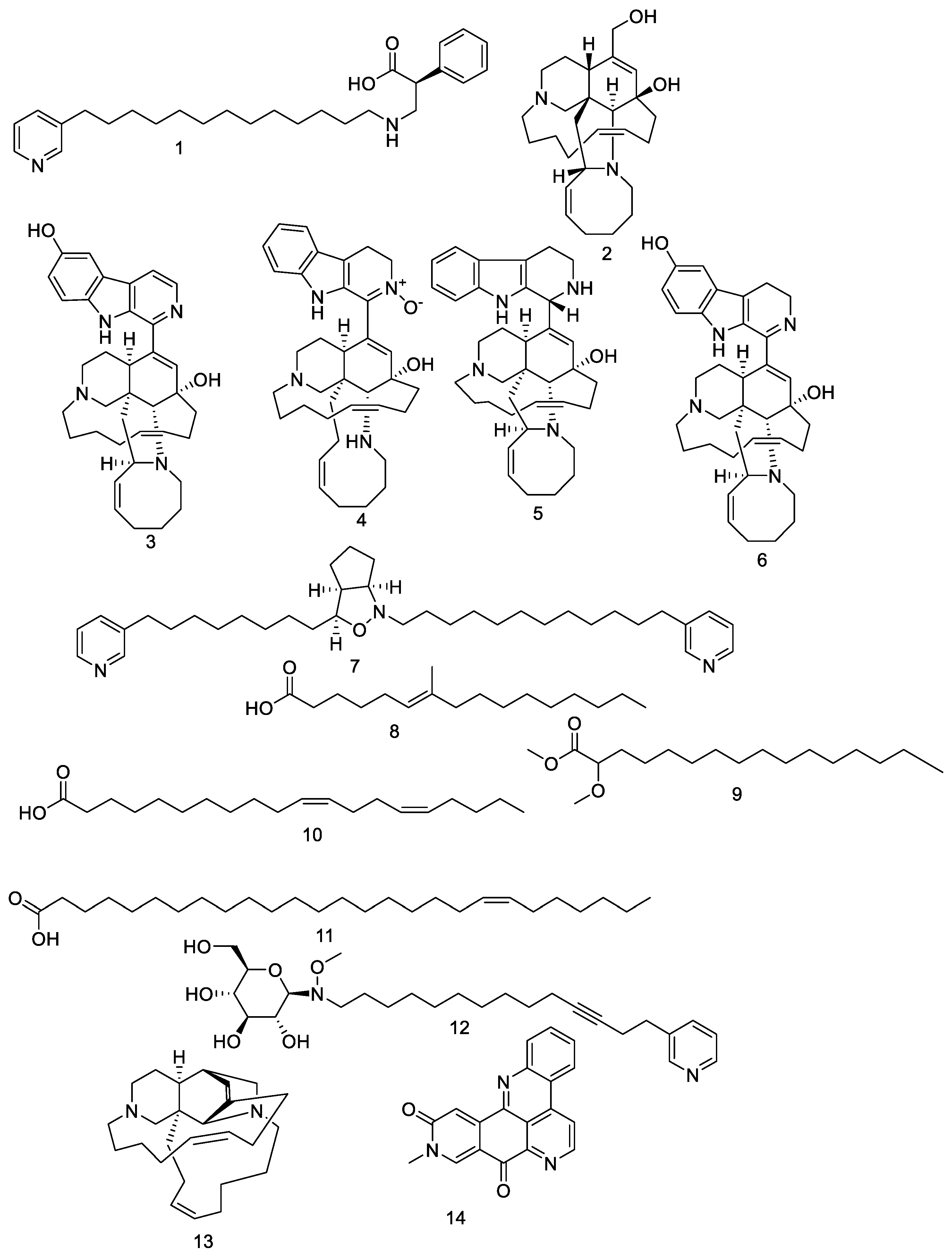
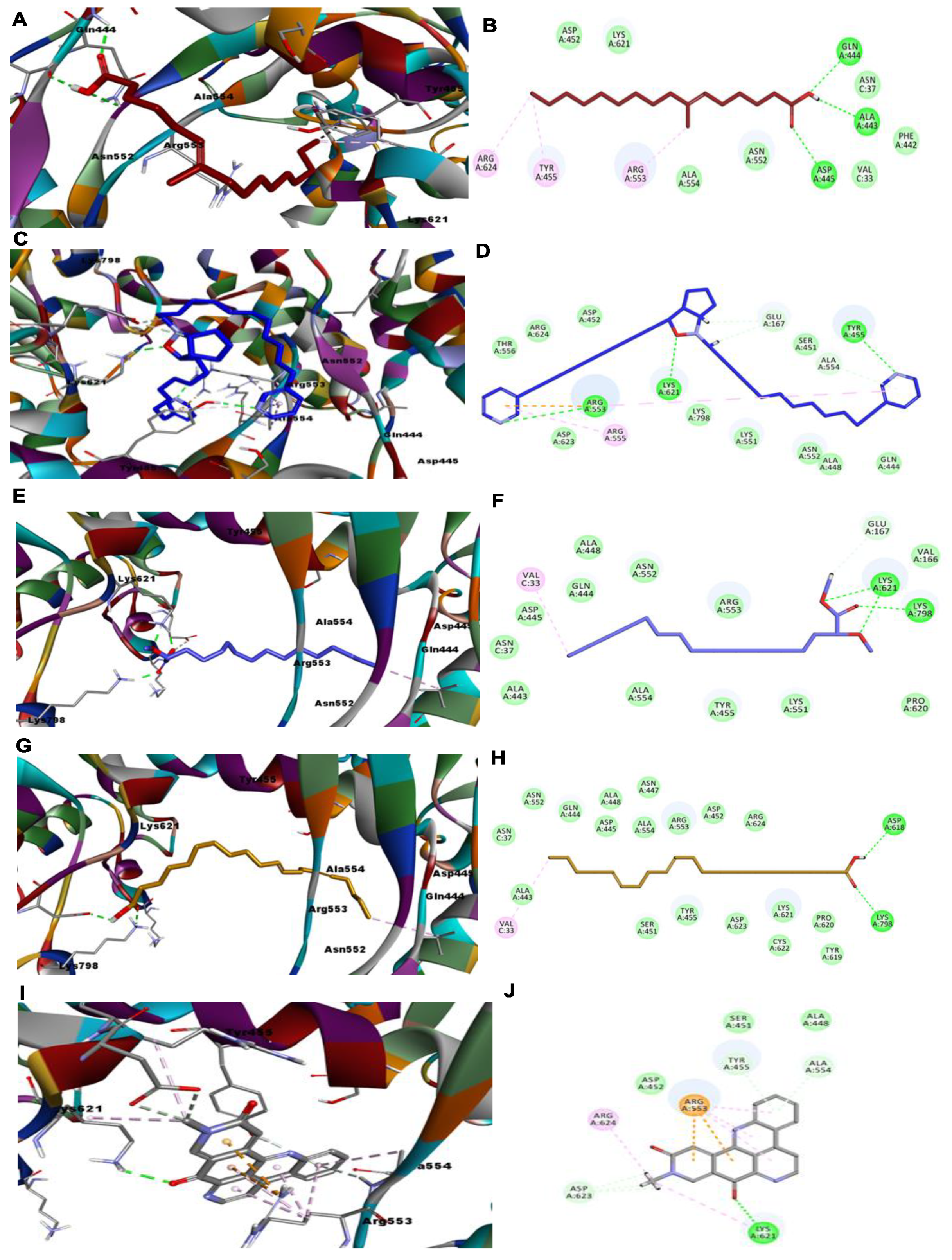
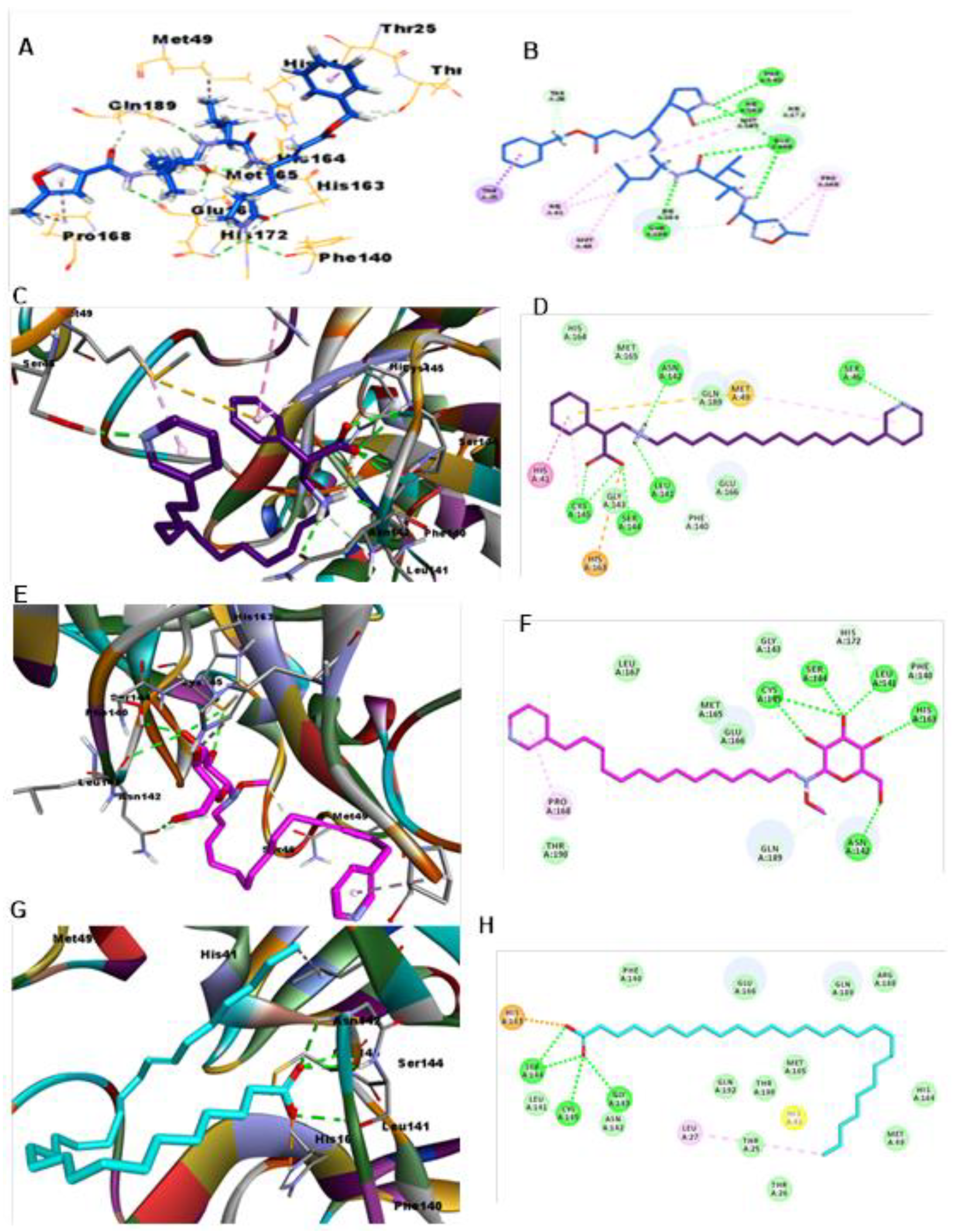
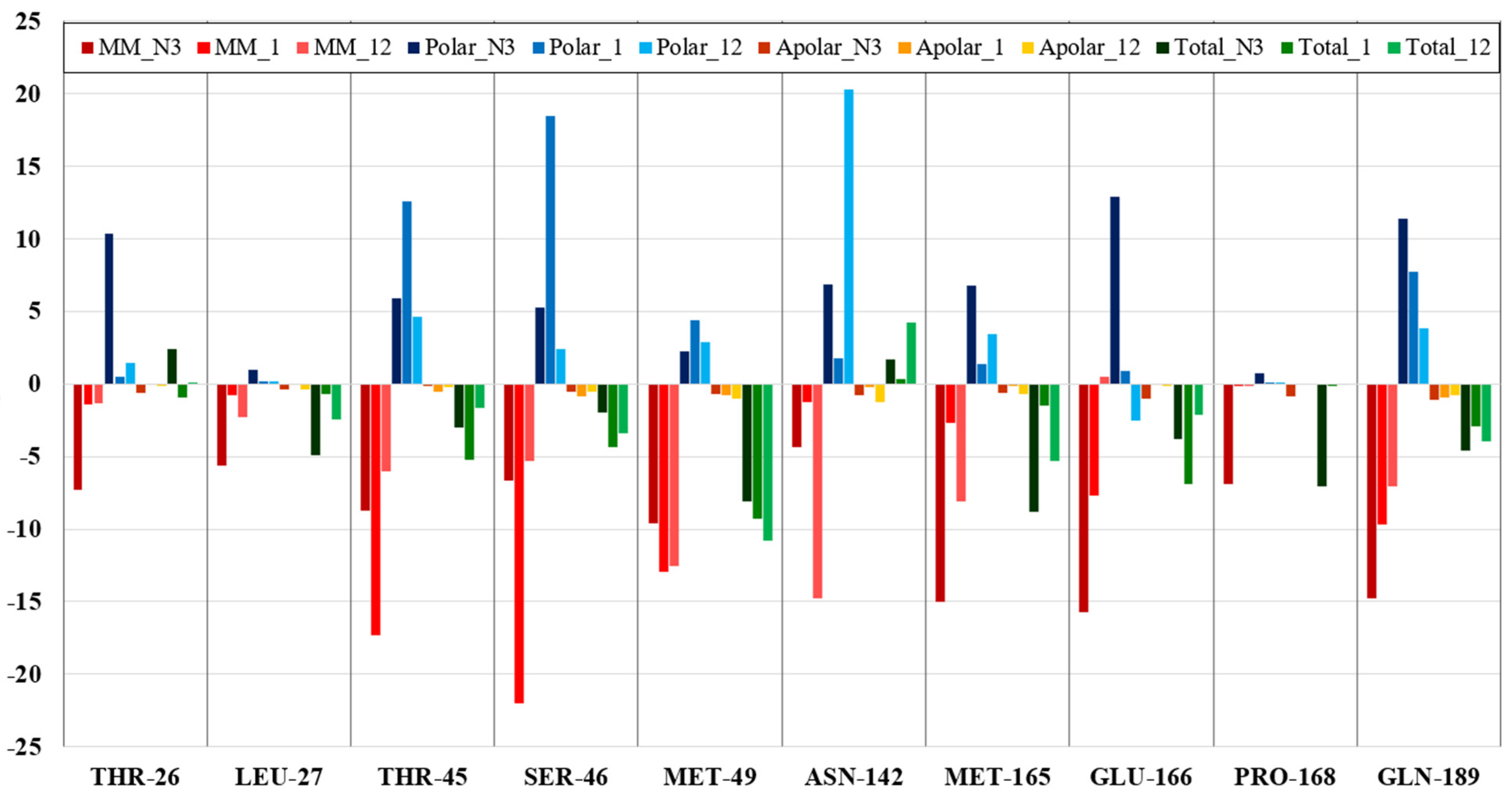
3. Results and Discussion
3.1. RdRp
3.2. CL Protease
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kandeel, M.; Al-Nazawi, M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020, 251, 117627. [Google Scholar] [CrossRef]
- Ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Yu, R.; Chen, L.; Lan, R.; Shen, R.; Li, P. Computational screening of antagonist against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int. J. Antimicrob. Agents 2020, 56, 106012. [Google Scholar] [CrossRef]
- Zahran, E.M.; Albohy, A.; Khalil, A.; Ibrahim, A.H.; Ahmed, H.A.; El-Hossary, E.M.; Bringmann, G.; Abdelmohsen, U.R. Bioactivity Potential of Marine Natural Products from Scleractinia-Associated Microbes and In Silico Anti-SARS-COV-2 Evaluation. Mar. Drugs 2020, 18, 645. [Google Scholar] [CrossRef]
- El-Hawary, S.S.; Rabeh, M.A.; Raey, M.A.E.; El-Kadder, E.M.A.; Sobeh, M.; Abdelmohsen, U.R.; Albohy, A.; Andrianov, A.M.; Bosko, I.P.; Al-Sanea, M.M.; et al. Metabolomic profiling of three Araucaria species, and their possible potential role against COVID-19. J. Biomol. Struct. Dyn. 2021, 1–13. [Google Scholar] [CrossRef]
- El-Hawary, S.S.; Mohammed, R.; Bahr, H.S.; Attia, E.Z.; El-Katatny, M.H.; Abelyan, N.; Al-Sanea, M.M.; Moawad, A.S.; Abdelmohsen, U.R. Soybean-associated endophytic fungi as potential source for anti-COVID-19 metabolites supported by docking analysis. J. Appl. Microbiol. 2021. [Google Scholar] [CrossRef]
- Shady, N.H.; Youssif, K.A.; Sayed, A.M.; Belbahri, L.; Oszako, T.; Hassan, H.M.; Abdelmohsen, U.R. Sterols and Triterpenes: Antiviral Potential Supported by In-Silico Analysis. Plants 2020, 10, 41. [Google Scholar] [CrossRef]
- Sayed, A.M.; Alhadrami, H.A.; El-Gendy, A.O.; Shamikh, Y.I.; Belbahri, L.; Hassan, H.M.; Abdelmohsen, U.R.; Rateb, M.E. Microbial Natural Products as Potential Inhibitors of SARS-CoV-2 Main Protease (M(pro)). Microorganisms 2020, 8, 970. [Google Scholar] [CrossRef]
- Hassan, H.A.; Abdelmohsen, U.R.; Aly, O.M.; Desoukey, S.Y.; Mohamed, K.M.; Kamel, M.S. Potential of Ficus microcarpa metabolites against SARS-CoV-2 main protease supported by docking studies. Nat. Prod. Res. 2020, 1–5. [Google Scholar] [CrossRef]
- Sagar, S.; Kaur, M.; Minneman, K.P. Antiviral lead compounds from marine sponges. Mar. Drugs 2010, 8, 2619–2638. [Google Scholar] [CrossRef] [Green Version]
- Mayer, A.; Guerrero, A.J.; Rodríguez, A.D.; Taglialatela-Scafati, O.; Nakamura, F.; Fusetani, N. Marine pharmacology in 2014–2015: Marine compounds with antibacterial, antidiabetic, antifungal, anti-inflammatory, antiprotozoal, antituberculosis, antiviral, and anthelmintic activities; affecting the immune and nervous systems, and other miscellaneous mechanisms of action. Mar. Drugs 2020, 18, 5. [Google Scholar]
- Abdelmohsen, U.R.; Balasubramanian, S.; Oelschlaeger, T.A.; Grkovic, T.; Pham, N.B.; Quinn, R.J.; Hentschel, U. Potential of marine natural products against drug-resistant fungal, viral, and parasitic infections. Lancet Infect. Dis. 2017, 17, e30–e41. [Google Scholar] [CrossRef]
- Abdelaleem, E.R.; Samy, M.N.; Desoukey, S.Y.; Liu, M.; Quinn, R.J.; Abdelmohsen, U.R. Marine natural products from sponges (Porifera) of the order Dictyoceratida (2013 to 2019); a promising source for drug discovery. RSC Adv. 2020, 10, 34959–34976. [Google Scholar] [CrossRef]
- Shady, N.H.; Fouad, M.A.; Kamel, M.S.; Schirmeister, T.; Abdelmohsen, U.R. Natural product repertoire of the Genus Amphimedon. Mar. Drugs 2019, 17, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shady, N.H.; Elfakharany, Z.; Salem, M.A.; Ahmed, S.; Fouad, M.A.; Kamel, M.S.; Krischke, M.; Mueller, M.J.; Abdelmohsen, U.R. Dereplication Analysis and Antitrypanosomal Potential of the Red Sea Sponge Amphimedon sp. Supported by Molecular Modelling. Rev. Bras. Farmacogn. 2020, 30, 290–294. [Google Scholar] [CrossRef]
- Shady, N.H.; Khattab, A.R.; Ahmed, S.; Liu, M.; Quinn, R.J.; Fouad, M.A.; Kamel, M.S.; Muhsinah, A.B.; Krischke, M.; Mueller, M.J.; et al. Hepatitis C Virus NS3 Protease and Helicase Inhibitors from Red Sea Sponge (Amphimedon) Species in Green Synthesized Silver Nanoparticles Assisted by in Silico Modeling and Metabolic Profiling. Int. J. Nanomed. 2020, 15, 3377–3389. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L. Discovery of a novel coronavirus associated with the recent pneumonia outbreak in humans and its potential bat origin. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, L.; Duan, Y.; Yu, J.; Wang, L.; Yang, K.; Liu, F.; You, T.; Liu, X.; Yang, X. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar]
- Abou-Zied, H.A.; Youssif, B.G.; Mohamed, M.F.; Hayallah, A.M.; Abdel-Aziz, M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 2019, 89, 102997. [Google Scholar] [CrossRef]
- Ibrahim, T.S.; Sheha, T.A.; Abo-Dya, N.E.; AlAwadh, M.A.; Alhakamy, N.A.; Abdel-Samii, Z.K.; Panda, S.S.; Abuo-Rahma, G.E.-D.A.; Mohamed, M.F. Design, synthesis and anticancer activity of novel valproic acid conjugates with improved histone deacetylase (HDAC) inhibitory activity. Bioorg. Chem. 2020, 99, 103797. [Google Scholar] [CrossRef]
- Al-Sanea, M.M.; Gotina, L.; Mohamed, M.F.; Parambi, D.G.T.; Gomaa, H.A.; Mathew, B.; Youssif, B.G.; Alharbi, K.S.; Elsayed, Z.M.; Abdelgawad, M.A. Design, Synthesis and Biological Evaluation of New HDAC1 and HDAC2 Inhibitors Endowed with Ligustrazine as a Novel Cap Moiety. Drug Des. Dev. Ther. 2020, 14, 497–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, M.F.A.; Youssif, B.G.M.; Shaykoon, M.S.A.; Abdelrahman, M.H.; Elsadek, B.E.M.; Aboraia, A.S.; Abuo-Rahma, G.E.A. Utilization of tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidinone as a cap moiety in design of novel histone deacetylase inhibitors. Bioorg. Chem. 2019, 91, 103127. [Google Scholar] [CrossRef] [PubMed]
- Youssif, B.G.M.; Mohamed, M.F.A.; Al-Sanea, M.M.; Moustafa, A.H.; Abdelhamid, A.A.; Gomaa, H.A.M. Novel aryl carboximidamide and 3-aryl-1,2,4-oxadiazole analogues of naproxen as dual selective COX-2/15-LOX inhibitors: Design, synthesis and docking studies. Bioorg. Chem. 2019, 85, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://biosig.unimelb.edu.au/pkcsm/prediction (accessed on 17 February 2021).
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shady, N.H.; Hayallah, A.M.; Mohamed, M.F.A.; Ghoneim, M.M.; Chilingaryan, G.; Al-Sanea, M.M.; Fouad, M.A.; Kamel, M.S.; Abdelmohsen, U.R. Targeting 3CLpro and SARS-CoV-2 RdRp by Amphimedon sp. Metabolites: A Computational Study. Molecules 2021, 26, 3775. https://doi.org/10.3390/molecules26123775
Shady NH, Hayallah AM, Mohamed MFA, Ghoneim MM, Chilingaryan G, Al-Sanea MM, Fouad MA, Kamel MS, Abdelmohsen UR. Targeting 3CLpro and SARS-CoV-2 RdRp by Amphimedon sp. Metabolites: A Computational Study. Molecules. 2021; 26(12):3775. https://doi.org/10.3390/molecules26123775
Chicago/Turabian StyleShady, Nourhan Hisham, Alaa M. Hayallah, Mamdouh F. A. Mohamed, Mohammed M. Ghoneim, Garri Chilingaryan, Mohammad M. Al-Sanea, Mostafa A. Fouad, Mohamed Salah Kamel, and Usama Ramadan Abdelmohsen. 2021. "Targeting 3CLpro and SARS-CoV-2 RdRp by Amphimedon sp. Metabolites: A Computational Study" Molecules 26, no. 12: 3775. https://doi.org/10.3390/molecules26123775
APA StyleShady, N. H., Hayallah, A. M., Mohamed, M. F. A., Ghoneim, M. M., Chilingaryan, G., Al-Sanea, M. M., Fouad, M. A., Kamel, M. S., & Abdelmohsen, U. R. (2021). Targeting 3CLpro and SARS-CoV-2 RdRp by Amphimedon sp. Metabolites: A Computational Study. Molecules, 26(12), 3775. https://doi.org/10.3390/molecules26123775