
Production of Minor Ginsenosides C-K and C-Y from Naturally Occurring Major Ginsenosides Using Crude β-Glucosidase Preparation from Submerged Culture of Fomitella fraxinea

 , ,
, ,
Abstract
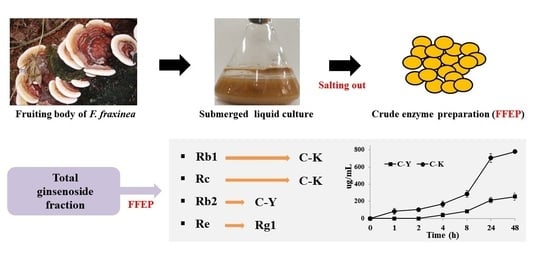
:
1. Introduction
2. Results and Discussion
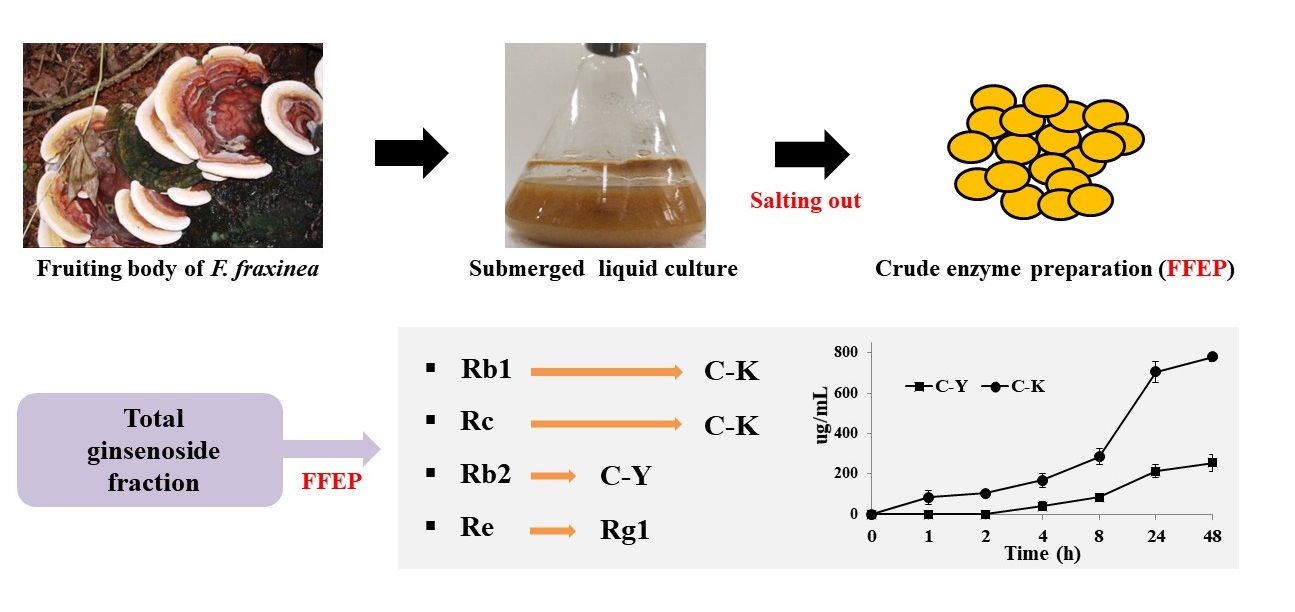
2.1. β-Glucosidase Activity in FFEP
2.2. Biotransformation of PPDG-F by FFEP
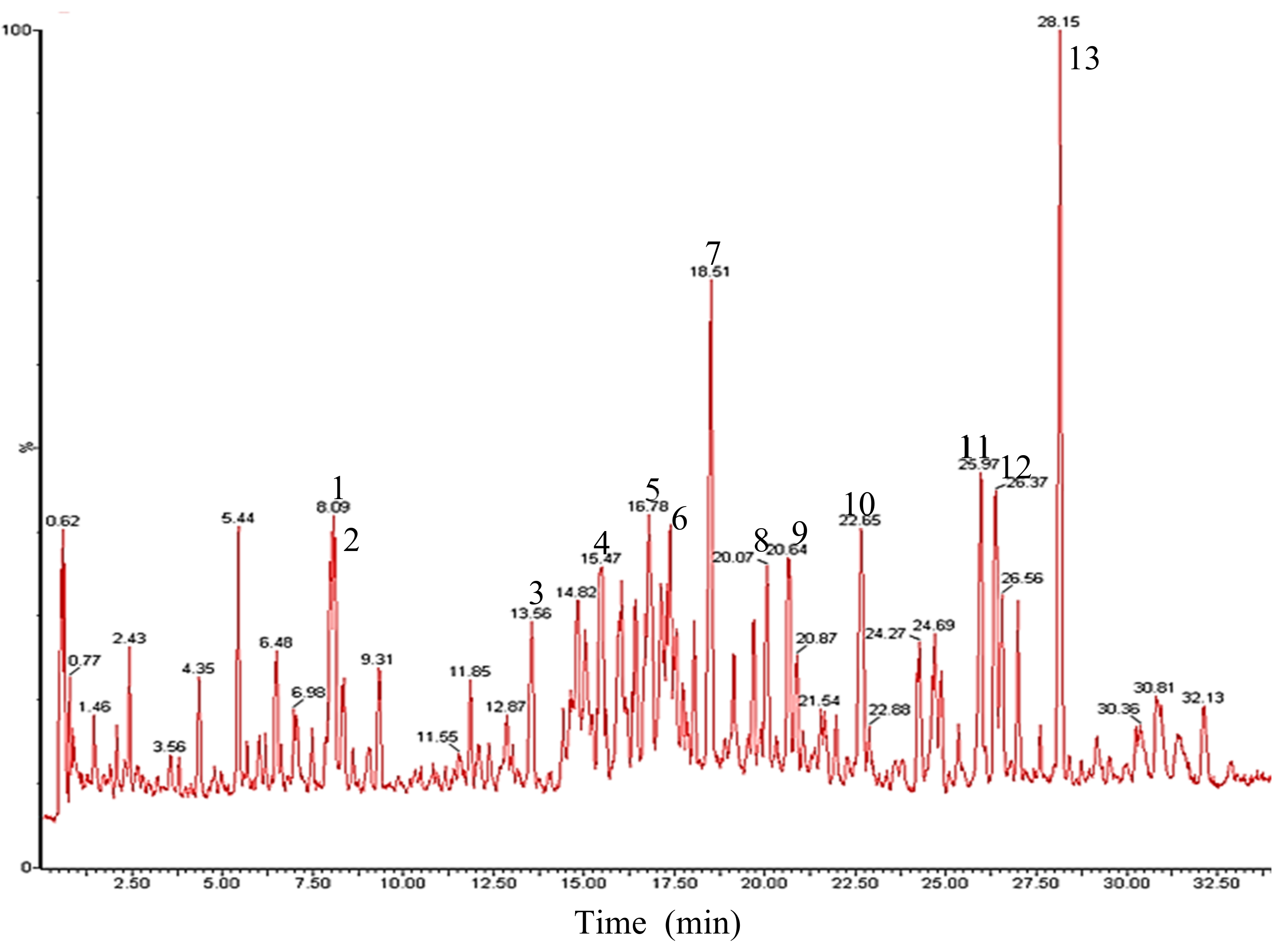
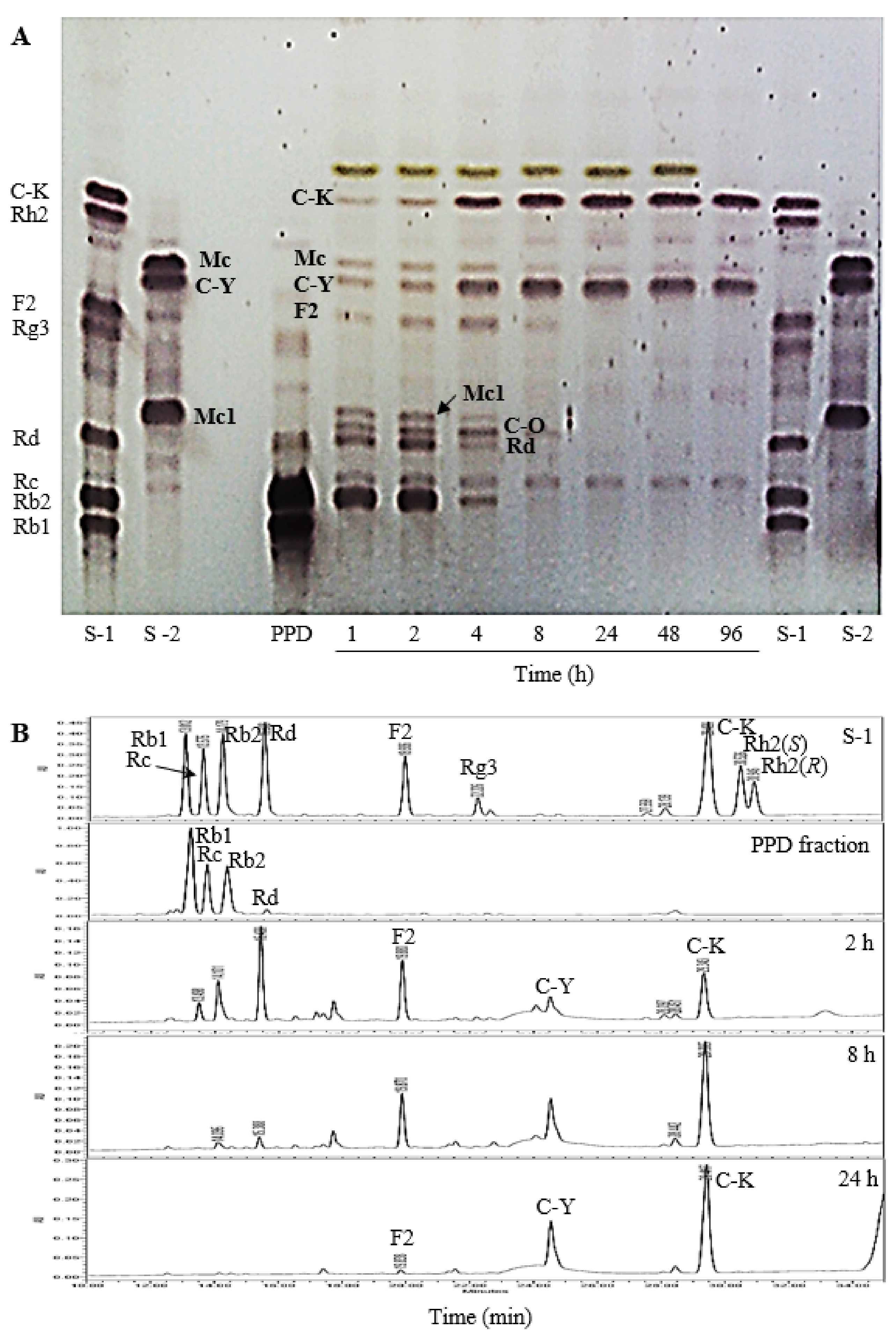
2.2.1. UPLC/ESI-Q-TOF-MS Analysis
2.2.2. The Effect of Reaction Time
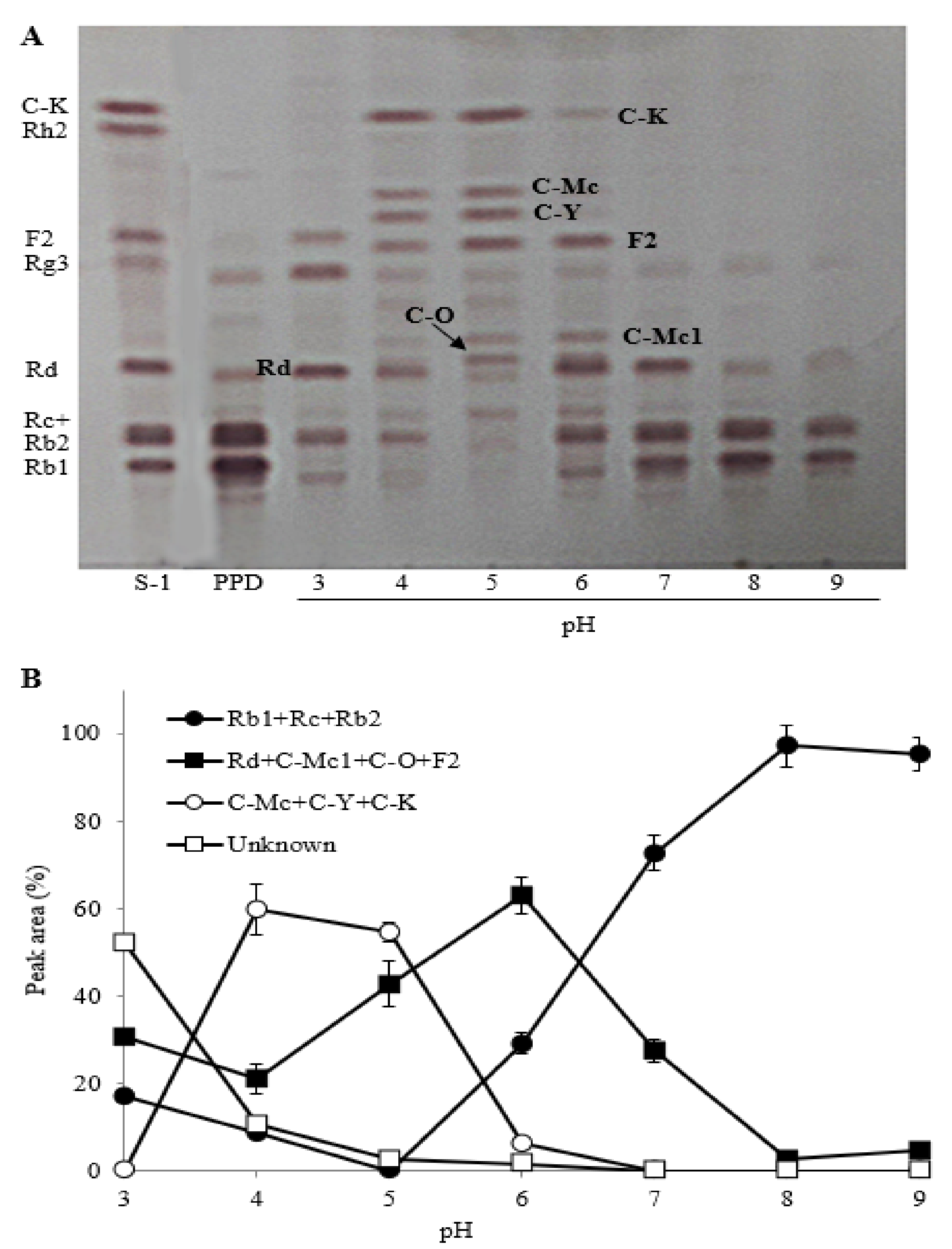
2.2.3. The Effect of Reaction pH
2.3. Biotransformation of TG-F by FFEP
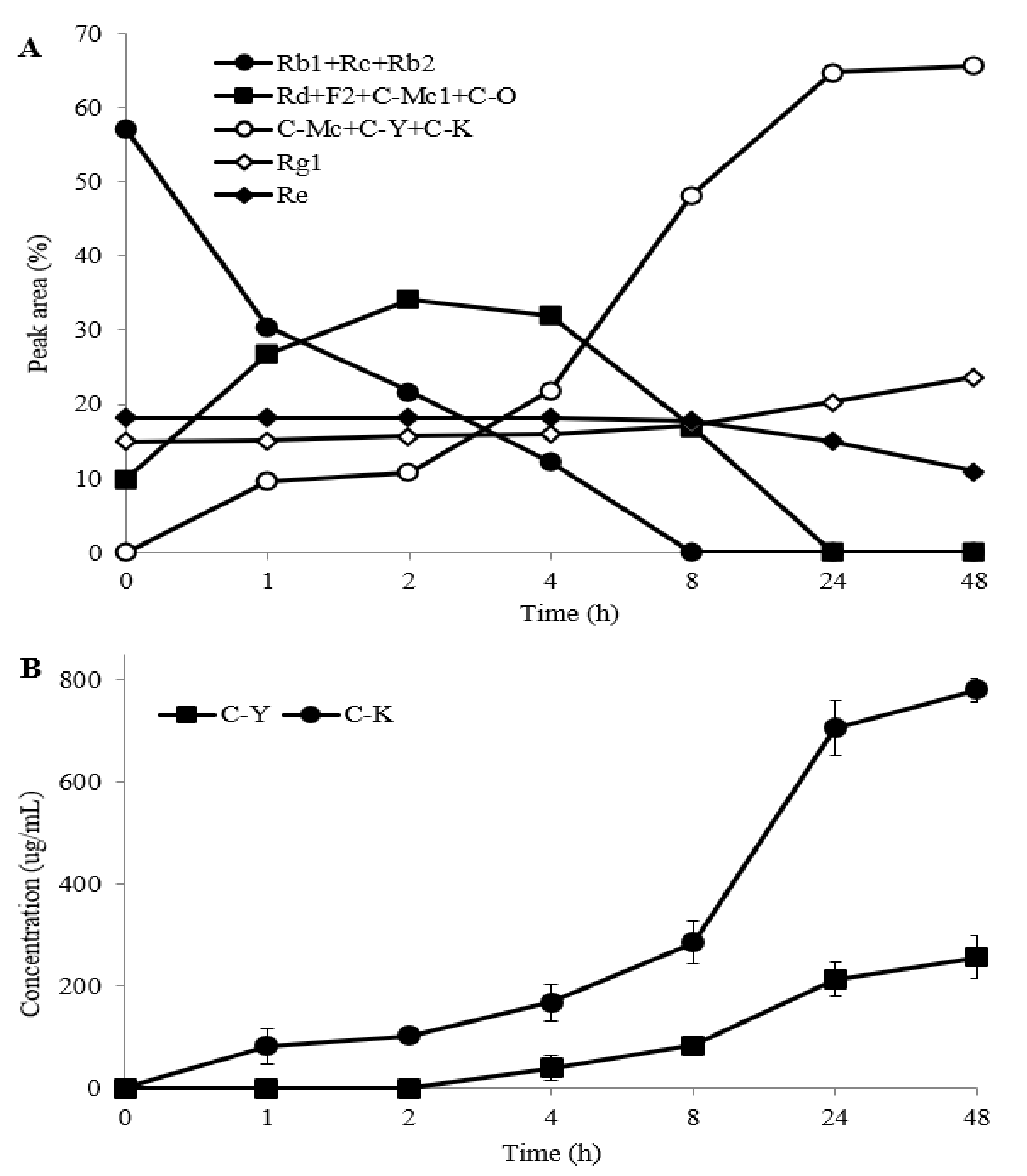
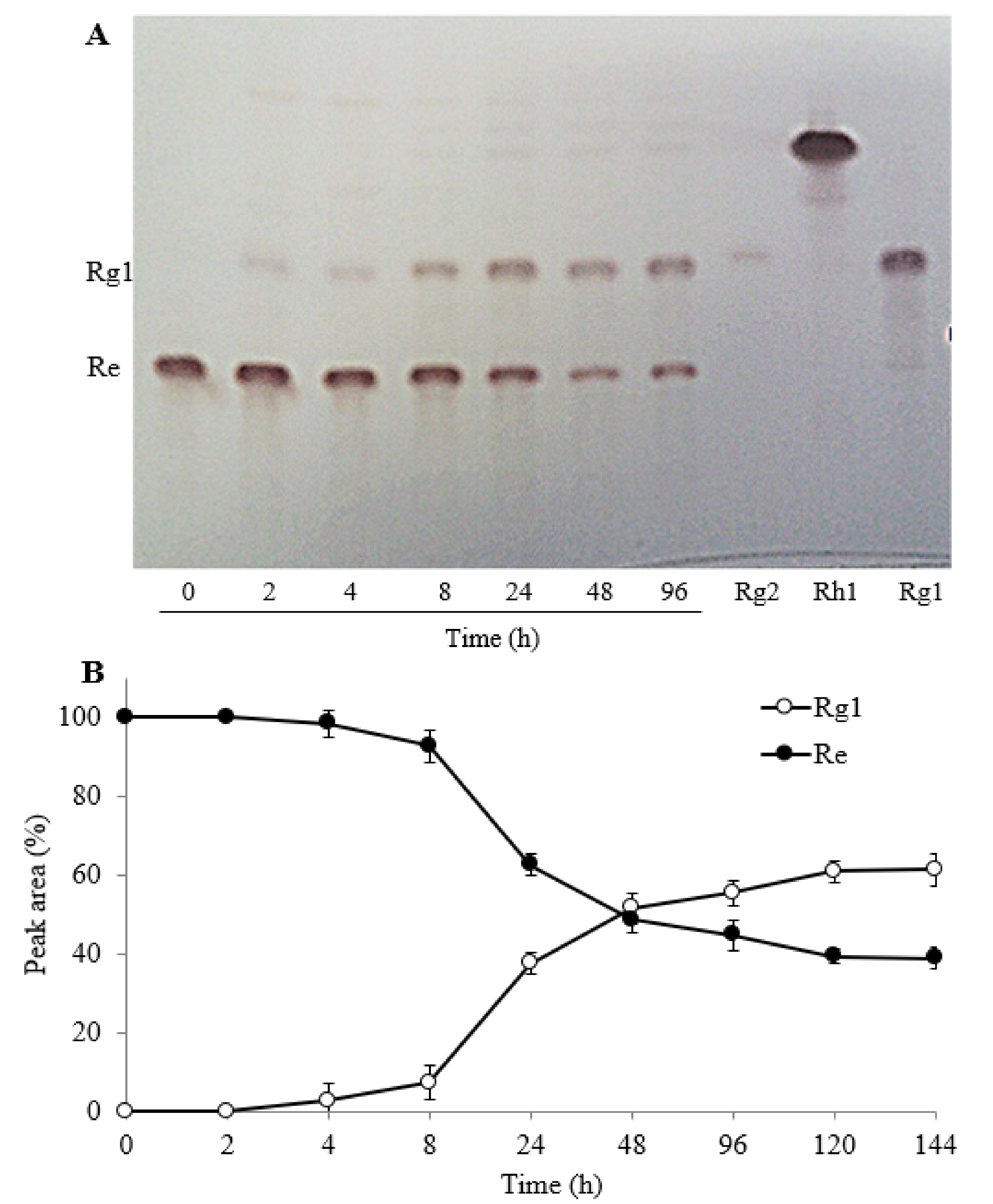
2.3.1. The Effect of Reaction Time
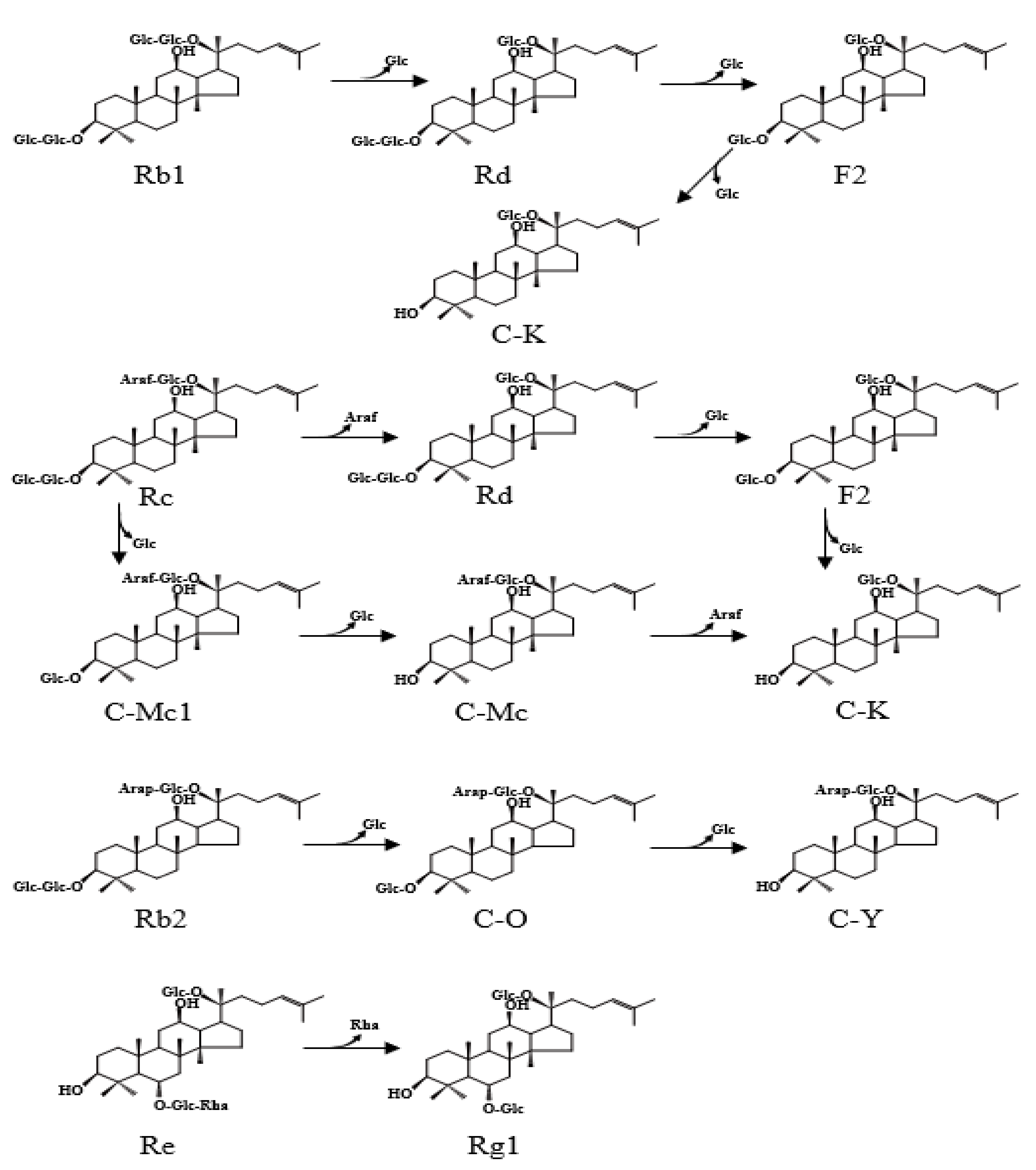
2.3.2. Biotransformation Pathway of Ginsenosides
3. Materials and Methods
3.1. Reagents
3.2. Preparation of Ginsenosides and Ginseng Extracts
3.3. Microorganism
3.4. Mushroom Cultivation
3.5. Preparation of Crude Enzyme
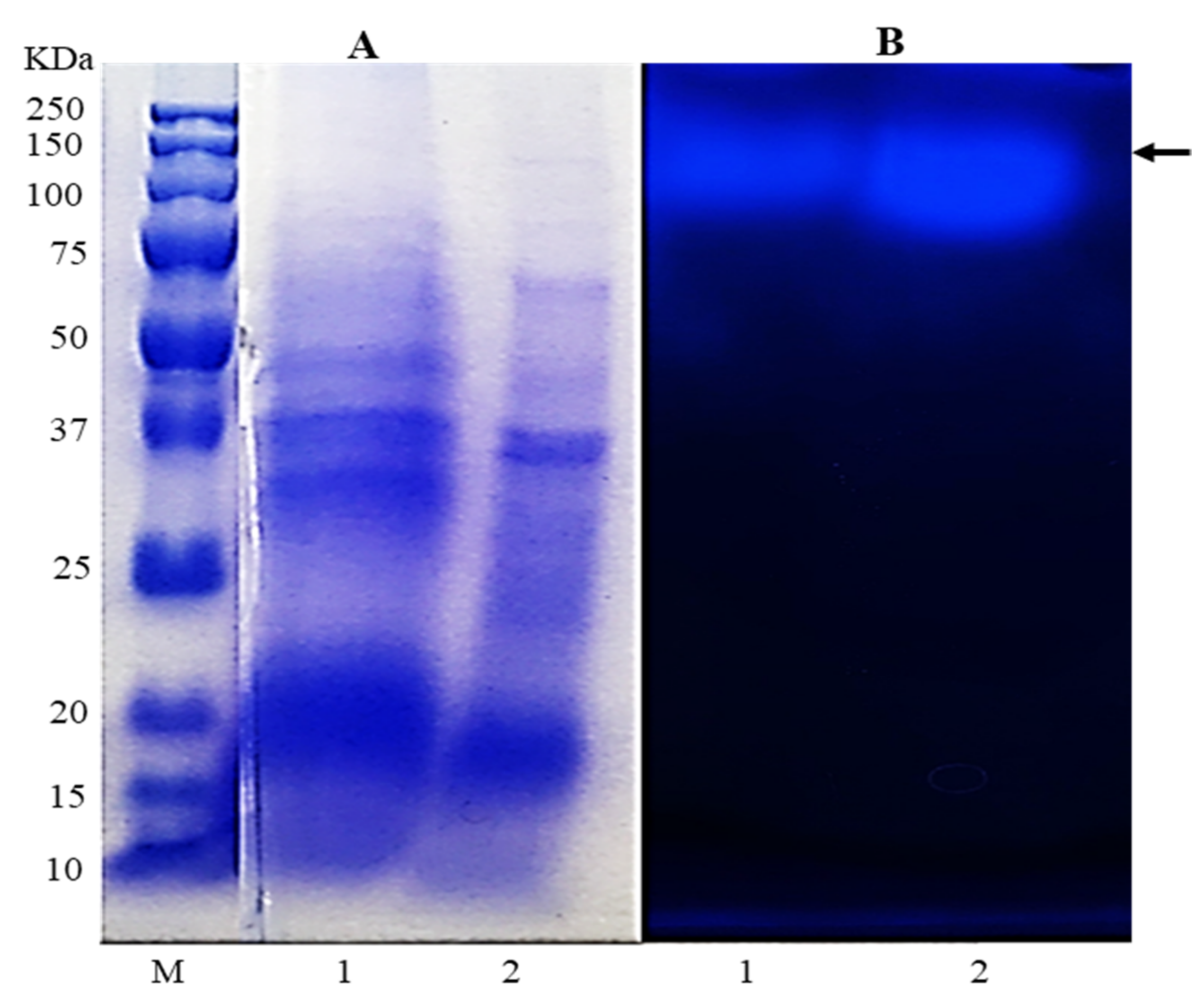
3.6. Enzyme Electrophoresis
3.7. Enzyme Activity and Protein Assays
3.8. Enzymatic Biotransformation
3.9. General Analytical Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Nocerino, E.; Amato, M.; Izzo, A.A. The aphrodisiac and adaptogenic properties of ginseng. Fitoterapia 2000, 71, S1–S5. [Google Scholar] [CrossRef]
- Radad, K.; Gille, G.; Liu, L.; Rausch, W.D. Use of ginseng in medicine with emphasis on neurodegenerative disorders. J. Pharmacol. Sci. 2006, 100, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.C.; Yang, J.; Lv, Y.H.; Chen, J.C.; Yin, F.; Huang, J.H.; Zheng, Q.S. A review of ginseng clinical trials registered in the WHO international clinical trials registry platform. BioMed. Res. Inter. 2018, 1843142. [Google Scholar] [CrossRef] [Green Version]
- Park, J.D.; Rhee, D.K.; Lee, Y.H. Biological activities and chemistry of saponins from Panax ginseng C. A. Meyer. Phytochem. Rev. 2005, 4, 159–175. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.T. Botanical characteristics, pharmacological effects and medicinal components of Korean Panax ginseng C. A. Meyer. Acta Pharmacol. Sin. 2008, 29, 1109–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.Y.; Park, J.H.; Kim, H.S.; Lee, C.Y.; Lee, H.J.; Kang, K.S.; Kim, C.E. Systems-level mechanisms of action of Panax ginseng: A network pharmacological approach. J. Ginseng Res. 2018, 42, 98–106. [Google Scholar] [CrossRef]
- Shergis, J.L.; Zhang, A.L.; Zhou, W.; Xue, C.C. Panax ginseng in randomised controlled trials: A systematic review. Phytother. Res. 2013, 27, 949–965. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H. Chemical diversity of Panax ginseng, Panax quinquifolium, and Panax notoginseng. J. Ginseng Res. 2012, 36, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ru, W.W.; Wang, D.L.; Xu, Y.P.; He, X.X.; Sun, Y.E.; Qian, L.Y.; Zhou, X.S.; Qin, Y.F. Chemical constituents and bioactivities of Panax ginseng (C. A. Mey.). Drug Discov. Ther. 2015, 9, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanada, S.; Kondo, N.; Shoji, J.; Tanaka, O.; Shibata, S. Studies on the saponins of ginseng. Ⅰ. Structures of ginsenoside-Ro, -Rb1, -Rb2, -Rc, and -Rd. Chem. Pharm. Bull. 1974, 22, 421–428. [Google Scholar] [CrossRef] [Green Version]
- Christensen, L.P. Ginsenosides chemistry, biosynthesis, analysis, and potential health effects. Adv Food Nutr. Res. 2009, 55, 1–99. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.K.; Kwon, S.W.; Park, J.H. Chemical diversity of ginseng saponins from Panax ginseng. J. Ginseng Res. 2015, 39, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H. Cardiovascular diseases and Panax ginseng: A review on molecular mechanisms and medical applications. J. Ginseng Res. 2012, 1, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Lee, H.J. Ginsenoside compound K: Insights into recent studies on pharmacokinetics and health-promoting activities. Biomolecules 2020, 10, 1028. [Google Scholar] [CrossRef]
- Tawab, M.A.; Bahr, U.; Karas, M.; Wurglics, M.; Schubert-Zsilavecz, M. Degradation of ginsenosides in humans after oral administration. Drug Metab. Dispos. 2003, 31, 1065–1071. [Google Scholar] [CrossRef] [Green Version]
- Kobashi, K.; Alkao, T. Relation of intestinal bacteria to pharmacological effects of glycosides. Biosci. Microflora 1997, 16, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, H. Proof of the mysterious efficacy of ginseng: Basic and clinical trials: Metabolic activation of ginsenoside: Deglycosylation by intestinal bacteria and esterification with fatty acid. J. Pharmacol. Sci. 2004, 95, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.F.; Fang, X.L.; Chen, D.F. Pharmacokinetics and bioavailability of ginsenoside Rb1 and Rg1 from Panax notoginseng in rats. J. Ethnopharmacol. 2003, 84, 187–192. [Google Scholar] [CrossRef]
- Won, H.J.; Kim, H.I.; Park, T.J.; Kim, H.M.; Jo, K.H.; Jeon, H.J.; Ha, S.J.; Hyun, J.M.; Jeong, A.R.; Kim, J.S.; et al. Non-clinical pharmacokinetic behavior of ginsenosides. J. Ginseng Res. 2019, 43, 354–360. [Google Scholar] [CrossRef]
- Shin, K.C.; Choi, H.Y.; Seo, M.J.; Oh, D.K. Compound K production from red ginseng extract by β-glycosidase from Sulfolobus solfataricus supplemented with α-L-arabinofuranosidase from Caldicellulosiruptor saccharolyticus. PLoS ONE 2015, 10, e0145876. [Google Scholar] [CrossRef]
- Yang, X.D.; Yang, Y.Y.; Ouyang, D.S.; Yang, G.P. A review of biotransformation and pharmacology of ginsenoside compound K. Fitoterapia 2015, 100, 208–220. [Google Scholar] [CrossRef]
- Oh, J.S.; Kim, J.S. Compound K derived from ginseng: Neuroprotection and cognitive improvement. Food Funct. 2016, 7, 4506–4515. [Google Scholar] [CrossRef]
- Kim, E.H.; Kim, W.N. An insight into ginsenoside metabolite compound K as a potential tool for skin disorder. Evid. Based Complement. Altern. Med. 2018, 8075870. [Google Scholar] [CrossRef]
- Han, B.H.; Park, M.H.; Han, Y.N.; Woo, L.K.; Sankawa, U.; Yahara, S.; Tanaka, O. Degradation of ginseng saponins under mild acidic conditions. Planta Med. 1982, 44, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.M.; Xu, F.X.; Li, Y.J.; Xi, X.Z.; Cui, X.W.; Han, C.C.; Zhang, X.L. Study on ttransformation of ginsenosides in different methods. BioMed. Res. Inter. 2017, 8601027. [Google Scholar] [CrossRef] [Green Version]
- Park, C.S.; Yoo, M.H.; Noh, K.H.; Oh, D.K. Biotransformation of ginsenosides by hydrolyzing the sugar moieties of ginsenosides using microbial glycosidases. Appl. Microbiol. Biotechnol. 2010, 87, 9–19. [Google Scholar] [CrossRef]
- He, W.N.; Wang, J.; Zhang, L.X.; Liu, Z.H. Biotransformation of ginsenosides and their aglycones. Inter. J. Biomed. Pharm. Sci. 2012, 6, 45–55. [Google Scholar]
- Jin, F.; Yu, H.; Fu, Y.; An, D.S.; Im, W.T.; Lee, S.T.; da Silva, J.A.T. Biotransformation of ginsenosides (ginseng saponins). Inter. J. Biomed. Pharm. Sci. 2012, 6, 33–44. [Google Scholar]
- Baldrian, P.; Valášková, V. Degradation of cellulose by basiomycetous fungi. FEBS Microbiol. Rev. 2008, 32, 501–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mfombep, P.M.; Senwo, Z.N.; Isikhuemhen, O.S. Enzymatic activites and kinetic properties of β-glucosidase from selected white rot fungi. Adv. Biol. Chem. 2013, 3, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, J.; Yoon, M.S.; Kim, M.J.; Ryu, N.S.; Song, Y.E.; Kim, Y.H.; Kim, M.K. Purification and characterization of a novel ginsenoside Rc-hydrolyzing β-glucosidase from Armillaria mellea mycelia. AMB Expr. 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Upadhyaya, J.; Yoon, M.S.; Ryu, N.S.; Song, Y.E.; Park, H.W.; Kim, Y.H.; Kim, M.K. Highly regioselective biotransformation of ginsenoside Rb2 into compound Y and compound K by β-glycosidase purified from Armillaria mellea mycelia. J. Ginseng Res. 2017, 42, 504–511. [Google Scholar] [CrossRef]
- Manavalan, T.; Manavalan, A.; Heese, K. Characterization of lignocellulolytic enzymes from white-rot fungi. Curr. Microbiol. 2015, 70, 485–498. [Google Scholar] [CrossRef]
- Bae, S.H.; Lee, H.S.; Kim, M.R.; Kim, S.Y.; Kim, J.M.; Suh, H.J. Changes of ginsenoside content by mushroom mycelial fermentation in red ginseng extract. J. Ginseng Res. 2011, 35, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Hsu, B.Y.; Lu, T.J.; Chen, C.H.; Wang, S.J.; Hwang, L.S. Biotransformation of ginsenoside Rd in the ginseng extraction residue by fermentation with lingzhi (Ganoderma lucidum). Food Chem. 2013, 141, 4186–4193. [Google Scholar] [CrossRef]
- Ryu, J.S.; Lee, H.J.; Bae, S.H.; Kim, S.Y.; Park, Y.; Suh, H.J.; Jeong, Y.H. The bioavailability of red ginseng extract fermented by Phellinus linteus. J. Ginseng Res. 2013, 37, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, Y.; Mishra, S.; Bisaria, V. Microbial β-glucosidases: Cloning, properties, and applications. Crit. Rev. Biotechnol. 2002, 22, 375–407. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Xu, S.J.; Li, X.N.; Yan, J.H.; Liu, L. Profiling the ginsenosides of three ginseng products by Lc-Q-Tof/Ms. J. Food Sci. 2013, 78, C653–C659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Li, S.L.; Zhang, H.; Wang, Y.; Zhao, Z.L.; Chen, S.L.; Xu, H.X. Holistic quality evaluation of commercial white and red ginseng using a UPLC-QTOF-MS/MS-based metabolomics approach. J. Pharm. Biomed. Anal. 2012, 62, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.R.; Suzuki, Y.; Choi, K.J.; Kim, Y.H. Enzymatic preparation of genuine prosapogenin, 20(S)-ginsenoside Rh1, from ginsenosides Re and Rg1. Biosci. Biotechnol. Biochem. 2000, 64, 2739–2743. [Google Scholar] [CrossRef] [Green Version]
- Chi, H.; Ji, G.E. Transformation of ginsenosides Rb1 and Re from Panax ginseng by food microorganisms. Biotechnol. Lett. 2005, 27, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak no. | RT (min) | Identification | [M − H]− | [M − H + HCOOH]− | Molecular Formula | |
|---|---|---|---|---|---|---|
| Theoretical Mass | Detected Mass | |||||
| 1 | 8.09 | G-Rg1 | 799.4845 | 799.7836 | 845.7795 | C42H72O14 |
| 2 | 8.11 | G-Re | 945.5248 | 945.8526 | 991.8694 | C48H82O18 |
| 3 | 13.56 | G-Rf | 799.4815 | 799.7791 | 845.7988 | C42H72O14 |
| 4 | 15.47 | G-Rh1 | 637.4316 | 637.4324 | 683.1782 | C36H62O9 |
| 5 | 16.78 | G-Rc | 1077.5781 | 1077.9037 | 1123.9282 | C53H90O22 |
| 6 | 17.31 | G-Rb2 | 1077.5683 | 1077.9022 | 1123.9231 | C53H90O22 |
| 7 | 18.51 | G-Rd | 945.5248 | 945.8588 | 991.8710 | C48H82O18 |
| 8 | 20.07 | C-Mc1 | 915.5396 | 915.8387 | 961.8601 | C47H80O17 |
| 9 | 20.64 | C-O | 915.7124 | 915.8281 | 961.8512 | C47H80O17 |
| 10 | 22.65 | G-F2 | 783.4895 | 783.7744 | 829.7908 | C42H72O13 |
| 11 | 25.97 | C-Mc | 753.4844 | 753.7594 | 799.7759 | C41H70O12 |
| 12 | 26.37 | C-Y | 753.4789 | 753.7576 | 799.7819 | C41H70O12 |
| 13 | 28.15 | C-K | 621.4366 | 621.6896 | 667.7121 | C36H62O8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.-W.; Lee, W.-J.; Gebru, Y.A.; Upadhyaya, J.; Ko, S.-R.; Kim, Y.-H.; Kim, M.-K. Production of Minor Ginsenosides C-K and C-Y from Naturally Occurring Major Ginsenosides Using Crude β-Glucosidase Preparation from Submerged Culture of Fomitella fraxinea. Molecules 2021, 26, 4820. https://doi.org/10.3390/molecules26164820
Kim D-W, Lee W-J, Gebru YA, Upadhyaya J, Ko S-R, Kim Y-H, Kim M-K. Production of Minor Ginsenosides C-K and C-Y from Naturally Occurring Major Ginsenosides Using Crude β-Glucosidase Preparation from Submerged Culture of Fomitella fraxinea. Molecules. 2021; 26(16):4820. https://doi.org/10.3390/molecules26164820
Chicago/Turabian StyleKim, Dae-Woon, Won-Jae Lee, Yoseph Asmelash Gebru, Jitendra Upadhyaya, Sung-Ryong Ko, Young-Hoi Kim, and Myung-Kon Kim. 2021. "Production of Minor Ginsenosides C-K and C-Y from Naturally Occurring Major Ginsenosides Using Crude β-Glucosidase Preparation from Submerged Culture of Fomitella fraxinea" Molecules 26, no. 16: 4820. https://doi.org/10.3390/molecules26164820
APA StyleKim, D. -W., Lee, W. -J., Gebru, Y. A., Upadhyaya, J., Ko, S. -R., Kim, Y. -H., & Kim, M. -K. (2021). Production of Minor Ginsenosides C-K and C-Y from Naturally Occurring Major Ginsenosides Using Crude β-Glucosidase Preparation from Submerged Culture of Fomitella fraxinea. Molecules, 26(16), 4820. https://doi.org/10.3390/molecules26164820