1. Introduction
High Performance Liquid chromatography (HPLC) has become one of the most powerful tools for the separation and determination of samples containing non-polar, moderately or strongly polar and ionic compounds, either simple species or complex high-molecular synthetic polymers or biopolymers. Non-ionic compounds are usually separated on the basis of differences in polarities, most often in reversed-phase (RP) systems with a non-polar stationary phase and a mixed aqueous–organic mobile phase. For the separation of moderately polar compounds, the conventional HPLC employs a polar adsorbent such as silica gel and a less polar mixed organic solvent mobile phase in organic normal-phase (NP) systems. However, strongly polar and partially ionized compounds are often weakly retained and poorly resolved in the RP systems and much too strongly (often irreversibly) retained in the NP systems. The issue was significantly alleviated by introduction of Hydrophilic Interaction Liquid Chromatography (HILIC) employing polar columns and mobile phases with high concentrations of polar organic solvents in water, i.e., aqueous normal-phase systems. Various polar columns may be suited for this purpose, including fully porous and core-shell silica gel, hybrid organic-silica, silica hydride, chemically bonded polar stationary phases with bonded cholesteryl, phenyl, nitrile, pentafluorophenyl propyl, diol, zwitterionic sulfobetaine, phosphorylcholine and other ligands. HILIC has been found especially useful in (not only) pharmaceutical, biomedical and clinical analysis.
Polar adsorbent surface has strong affinity towards water, which it takes up from aqueous–organic mobile phases. There is general agreement that the water adsorbed in concentrations exceeding the volume fraction in the bulk mobile phase is the main origin of the retention of polar compounds in HILIC [
1]. The computer molecular dynamics simulation studies indicate that the relative proportion of the amount of water contained in the pores of silica-based phases to the water concentration in the bulk mobile phase increases at low total water concentrations in the column [
2] This may be the reason why the LC separations are often irreproducible or fail in the mobile phases containing less than 2% water in acetonitrile. The water molecules close to the silica surface strongly adhere to the silanol groups by the hydrogen bonds. Three types of water molecules co-exist inside the 6–10 nm pores: free water molecules, “freezable” bound water, and bound water that does not freeze at the regular water freezing temperature [
3].
Because water is miscible at any proportion with acetonitrile, acetone, methanol or other polar organic solvents used in the HILIC mode, the adsorbed water forms a diffuse layer lacking fixed boundaries. The concentration of water progressively decreases from the polar solid surface towards the bulk organic-rich mobile phase outside (possibly, even partly inside) the pores of the stationary phase [
4]. The retention mechanism in HILIC depends on the polarity of the column material. The adsorbed water layer plays principal role in the retention mechanism.
In HPLC, the retention time is controlled by the ratio of the time spent by a solute in the stationary phase,
ts, to the time spent in the mobile phase,
tm, i.e., the retention factor,
k, which is equal to the ratio of the masses of the solute in the stationary,
ms, and in the mobile,
mm, phases, in the column.
k is directly proportional to the constant,
KD, characterizing the distribution of the solute between the stationary and the mobile phase:
The retention factor, k, depends on the nature of the solute, of the stationary and the mobile phases and on temperature, but is independent of the flow rate of the mobile phase, the dimensions of the column (provided equal density of packing, i.e., a constant phase ratio along the column). Hence, k is a fundamental parameter in the method development and optimization of HPLC separations and for measuring thermodynamic quantities of the retention process. Unfortunately, the retention factors calculated from the experimental retention data using Equation (1) do not provide reliable information on the presumed mechanism of retention, as several different mechanisms may contribute to the actual k.
Ideally, the equilibrium distribution of the sample compounds between the stationary and the mobile phase instantaneously re-establishes at any time in any part of the chromatographic bed. The changes in the partial molar Gibbs free energy, Δ
G, of the solute transfer from the mobile to the stationary phase control the thermodynamics of the chromatographic for strongly diluted samples:
R is the gas constant,
T is the temperature (in Kelvins) and
KD is the distribution (partition) constant, which gives the equilibrium ratio of the concentrations of the solute in the stationary,
cs, and in the mobile,
cm, phases, respectively [
5].
The proportionality constant Φ in Equation (1) is the phase ratio, i.e., the ratio of the volumes of the stationary,
Vs, and of the mobile,
Vm, phases in the column. From the Equation (2), it follows that:
tm and
Vm are also known as the column hold-up time and hold-up volume, respectively. The terms
t0, and
V0 are sometimes used instead of
tm and
Vm. The ratio of the column length,
L, to the column hold/up time,
tm, controls the linear velocity of the mobile phase along the column,
u.
F is the volume flow rate of the mobile phase, a simple conversion factor between the retention times and retention volumes [
6].
For correct determination of the retention factors, we must know the volumes of the stationary and mobile phases in the column, which determine the column phase ratio, Φ = Vs/Vm in Equation (1).
Simple estimation of the column hold-up time,
t0, as the time of the appearance of the first disturbance peak on the detector baseline may be subject to serious errors. A more sophisticated estimation involves setting the void volume equal to the elution volume of an “inert” compound, which neither taken up nor excluded from the solid phase (after the correction for extra-column volumes). The selection of a suitable marker compound is generally pragmatic and may not always yield the correct
Vm values. The marker selection is especially challenging in HILIC systems. Small polar molecules such as uracil or thiourea are often used as the column markers in reversed-phase chromatography, non-polar hydrocarbons such as benzene or toluene in the HILIC systems [
4]. However, the elution times of benzene and toluene on silica gel columns may slightly increase at 30% or more water in the mobile phase. This means that the water amount adsorbed close to the polar adsorbent surface depends on the water concentration in the bulk mobile phase [
7]. Even the retention time of a component of the mobile phase or of a monovalent salt may not provide accurate
Vm values [
8]. Integrating the plots of the retention times of the perturbation peak from 0 to 100 % of the organic modifier [
9] is time consuming and the peak is not always clearly apparent, or may be due to the impurities excluded before the real column hold-up time [
10]. Static methods of the determination of the column hold-up volume, such as the subsequent weighing of the column filled with two solvents of different densities [
8], do not account for the preferential solvation of the stationary phase. Linearization of the logarithmic net retention times of the members of a homologous series for the determination of
Vm is a lengthy approach and presumes validity of the ideal reversed-phase model [
11].
The assignment of the solvent adsorbed on the solid phase surface either to the stationary phase or to the mobile phase volume is still controversial [
12]. Further, the exact position of the boundary (dividing plane) between the bulk mobile phase and the liquid occluded on the stationary phase is difficult, if possible at all [
13]. The determination of the volume of the stationary phase is even more difficult in HILIC systems, because of the diffuse character of the stationary phase layer (water) in the inner pores of the column packing particles. According to the original Alpert model, the excess adsorbed water represents the stationary phase in HILIC [
1]. Dinh et al. [
14] presume that desorption and partitioning actually coexist as retention promoters for neutral solutes. Guo et al. [
15] proposed determination of the sorbed water layer as the difference between the elution volumes of toluene in the aqueous/organic mobile phase and in pure acetonitrile. The distribution constant in LC depends on the retention mode and—deliberately or unintentionally—leads to the phase definition by accepting a convention.
Convention 1 (classical): This is the standard convention used in the traditional determination of the retention factors in LC. The volume of the stationary phase, Vs, is inaccessible to a non-retained marker compound neither retained in nor excluded from the stationary phase. This means that Vs is equal to the empty column inner volume, Vcolumn, minus the total pore volume, VT (the inner pores, Vi, plus the inter-particle pores, V0):Vs = Vcolumn − Vi − V0. In HILIC, a small non-polar hydrocarbon molecule (benzene or toluene) is the most frequent Vm marker.
Convention 2 (inner pore): In the (idealized) size-exclusion chromatography (SEC), the solid particle skeleton, VSKEL, is inert and does not participate in the distribution process. The full volume of the inner pores contains the liquid stationary phase, Vs = Vi, which has the same composition as the mobile phase in the inter-particle volume (such as tetrahydrofuran), Vm = V0. The molecules of the solute distribute between Vi and V0, based on their size, which controls the part of the accessible pore volume. The elution volume of a high-molecular weight standard for which the inner pores are inaccessible, (e.g., polystyrene with Mr ≥ 106), estimates the volume of the mobile phase, Vm; the elution volume of a small hydrophobic molecule (benzene or toluene) estimates the volume of the sum of the inner and inter-particle pores, Vi + V0.
Convention 3 (adsorbed water): In HILIC—according to the original Alpert model—the sample distributes between the bulk, organic solvent-rich mobile phase and volume of water on the polar solid surface [
1]. Hence, in the HILIC systems, the
convention 2, setting the whole inner pore volume equal to the volume of the stationary phase in the column cannot be applied, as water (the liquid stationary phase) fills only a larger or a smaller part of the inner pore volume, in which it is diffused. The liquid in the inner pores differs from the bulk liquid mobile phase, contained in the inter-particle volume and in a part of the inner pores. The amount of the diffused water stationary-phase strongly depends on the composition of the aqueous–organic bulk mobile phase. The solid particle skeleton may contribute to the retention, mainly by the surface adsorption, and we can neglect its contribution to the retention at first approximation, so that we can understand the adsorbed diffuse layer of water as the stationary phase,
Vs =
Vex, and the remaining liquid inside the pores and in the inter-particle volume as the mobile phase,
Vm =
Vi +
V0 −
Vex. Hence, the differences between the phase ratios defined by the two conventions depend on the composition of the mobile phase and are more significant for columns with stronger water uptake.
3. Results and Discussion
For each column, the hold-up volume,
VM, was measured using
convention 1 as the elution volume of toluene in 100% and 95% acetonitrile, respectively. The interstitial volume in between the particles,
V0, was determined as the elution volume of the narrow-distribution high-molecular polystyrene standard with
Mr = 1,800,000 in 100% tetrahydrofuran. The total column porosity,
εT, was calculated from the volume of the empty column,
Vcolumn, and the column hold-up volume,
VM, (
εT =
VM/
Vcolumn), the interstitial column porosity as
ε0 =
V0/
Vcolumn. The inner porosity was obtained as the difference between the column total porosity and the interstitial porosity,
εi =
εT −
ε0, i.e., the pore volume inside the column was calculated as the difference between the hold-up volume and the interstitial volume,
Vi =
VM −
V0. The dimensions and pore characteristics of 18 columns studied are listed in
Table 1.
Vs defined by
convention 3 strongly depend on the composition of the aqueous–organic bulk mobile phase. These effects are described by the isotherms of water on the individual columns. We combined a dynamic frontal analysis method with Karl Fischer titration as a direct method of determination of water in the collected effluent fractions [
16]. The experimental isotherms of water adsorbed in the inner particle pores in excess of the bulk mobile phase concentration on the columns tested satisfactorily characterizes the Langmuir model, as shown in Equation (7) [
17]:
qi is the volume fraction of water contained in the pores of the stationary phase in excess of the water contained in the bulk mobile phase,
cm and
a is the distribution constant of water in the pores of the stationary phase at a very low
cm.
qs in Equation (6) gives the maximum (saturation) column adsorption capacity for water.
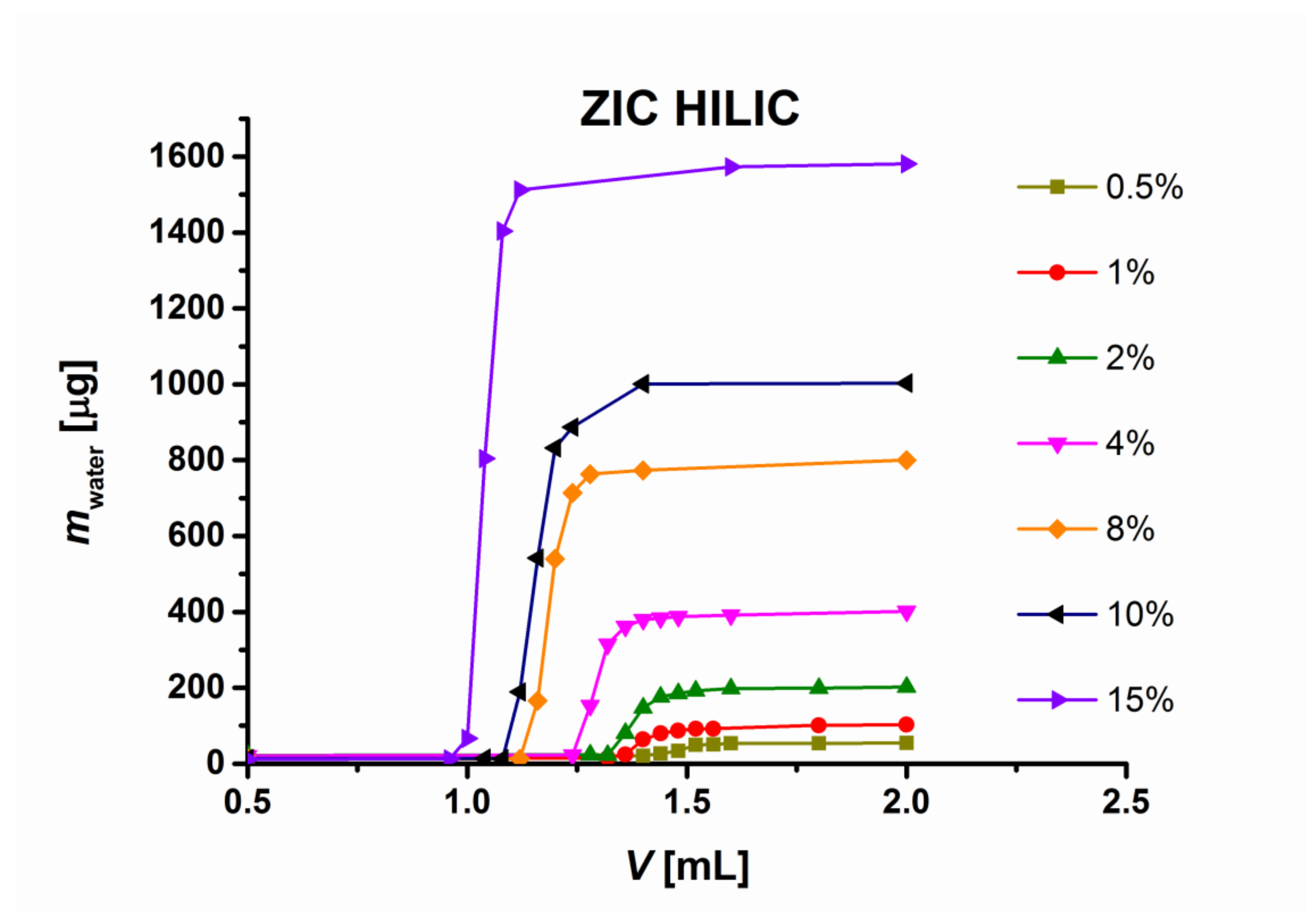
Figure 2 shows three examples of water isotherms on a zwitterionic ZIC-HILIC, a TSKgel amide and a hydrosilated Cogent Silica C columns [
4].
The adsorption isotherms show the excess volume of water (beyond the water concentration in the bulk mobile phase) retained in a column.
Table 2 shows large differences in the proportions of the inner pores occupied by the excess adsorbed water, calculated as
Vex =
qsatur·
V0,
Vex (from 7 to 129 µL) for the 18 columns tested. The water sorption saturation capacities (in excess of the water concentration in the bulk mobile phase),
qex and the corresponding excess volume of water contained in the adsorbed diffuse layer,
Vex. Even non-polar or slightly polar columns intended for reversed-phase LC applications (such as Cogent Cholesterol, Cogent Bidentate C18, Ascentis PhF5, Cogent Phenyl hydride) show a weak water adsorption. Due to a low affinity to water, hydrosilated silica stationary phases adsorb less than one monomolecular water layer equivalent (corresponding to 0.45 water monolayers for Cogent Silica C). Less than 9%
v/
v water in the mobile phase is sufficient to accomplish the full water saturation capacity of porous, core-shell, hybrid Ethylene Xbridge HILIC and hydrosilated silica gel columns, such as Cogent Silica C. On the other hand, the water adsorption isotherms on the ZIC HILIC and TSK gel amide 80 columns are shallow and even the mobile phases with 20% water/acetonitrile does not allow the achieving of full water saturation capacity (
Figure 2). Columns with bonded polar ligands (hydroxyl, diol, nitrile, pentafluorophenylpropyl, diol, zwitterionic sulphobetaine and phosphorylcholine), and TSK gel amide and amine, show stronger water adsorption in comparison to the bare silica. At full column saturation, the amount of the excess adsorbed water,
Vex, corresponds to 6–9 monomolecular equivalents of water layers,
Nw, for the ZIC cHILIC, and ZIC HILIC zwitterionic stationary phases, to 3–5 monomolecular water layer equivalents for the stationary phases with bonded hydroxyl groups (Luna HILIC, YMC Triart diol and Ascentis Express OH5) and to 1–2 water layer equivalents for the Xbridge HILIC and Atlantis HILIC columns [
14].
The
Table 2 and
Figure 3 show large differences in the proportion of the volume of the column inner pores filled with the excess water (100
εH2O/
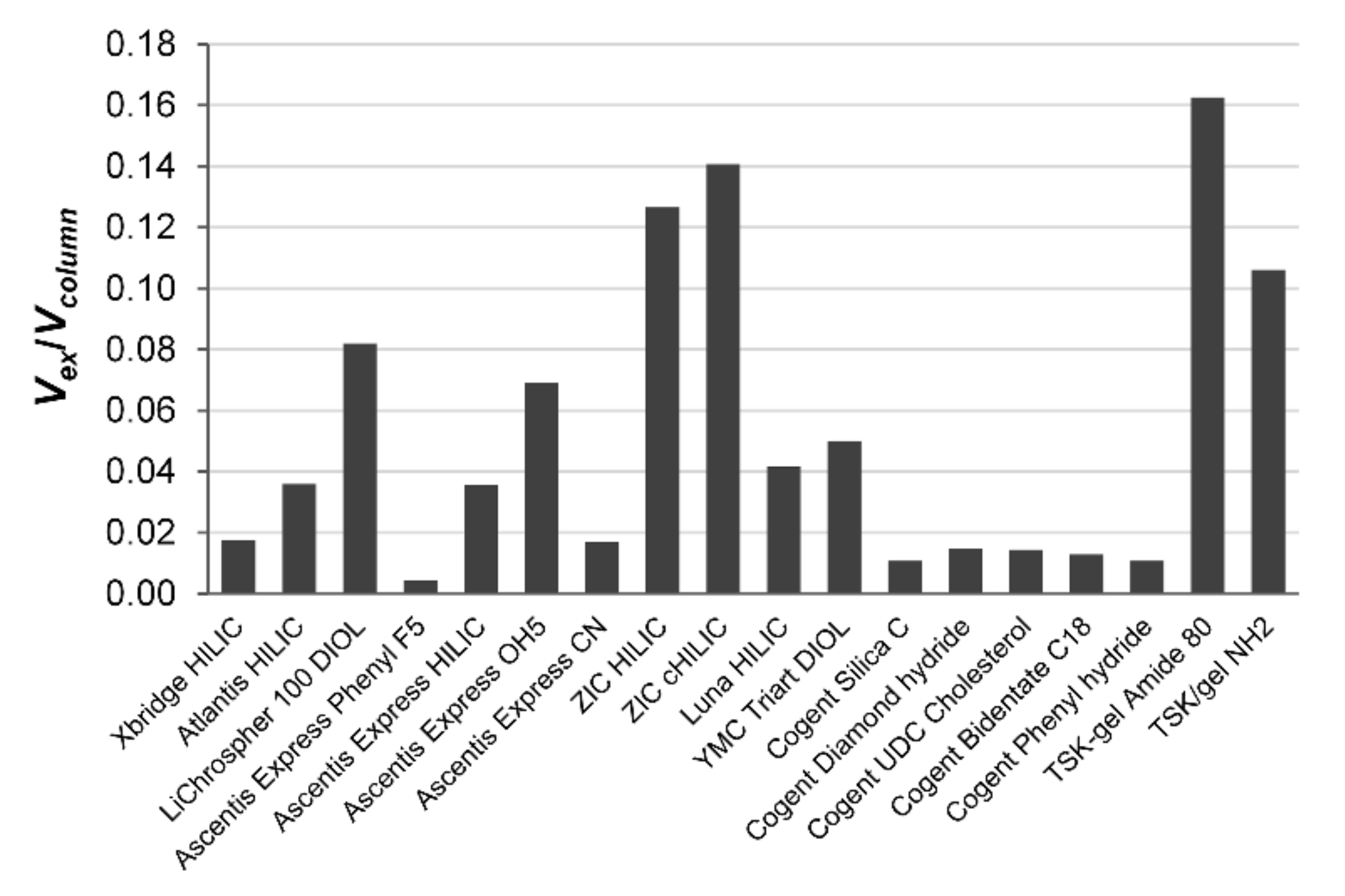
ε0) in aqueous acetonitrile containing 15% water. In the fused-core shell silica columns, the nonporous core occupies a significant part of the void column volume; the excess adsorbed water,
Vex, fills only 2.1% of the pore volume of the Ascentis Express Phenyl FS column, 8.5% of the Ascentis Express CN nitrile column, 1.7% of the Ascentis Express HILIC column and 3.5% of the Ascentis Express OH5 column. In the fully porous silica-based stationary phases containing –OH groups, LiChrospher 100 DIOL, YMC triart DIOL and Luna HILIC, the adsorbed excess water takes 2.4%, 15.1% and 11.3% pore volume, respectively. The hybrid organic-silica Xbridge HILIC and the Atlantis HILIC columns contain 5.3% and 9% adsorbed excess water, respectively, while the pore volume of less polar hydrosilated silica Cogent columns is filled by excess adsorbed water only to 2.7–5.4%. McCalley and Neue [
7] published similar results suggesting that 4–13% of the pore volume of a silica-bonded phase is occupied by a water-rich layer in 95–75% (
v/
v) aqueous acetonitrile. Maximum water uptake in the acetonitrile with 15% water is observed for zwitterionic stationary phases ZIC HILIC (32%) and ZIC cHILIC (20%), and for TSK-gel Amide 80 (51%) and TSK-gel NH2 (46%).
Table 2 lists the porosities characterizing the proportions of the column volume corresponding to the inner pores, the inter-particle space and the volume occupied by the adsorbed water. The pore distribution is illustrated by the pie diagrams in
Figure 4 for the ZIC HILIC, TSK Amide, Triart Diol, Luna HILIC and X-Bridge HILIC columns.
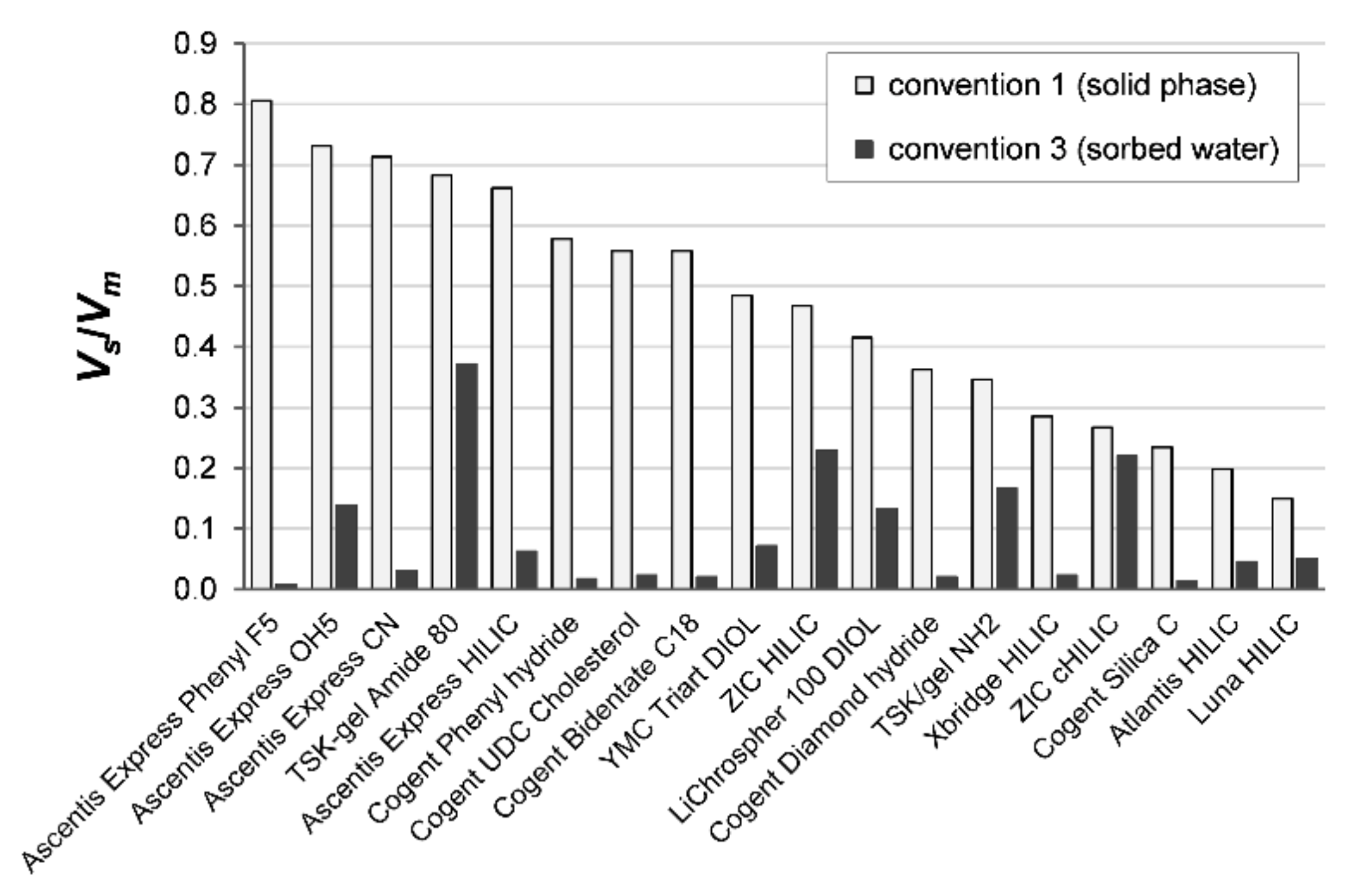
Figure 5 compares the volume phase ratios in the 18 columns according to the two conventions. There are more or less significant differences in the column volume ratios of the stationary and the mobile phases,
Vs/
Vm, according to
convention 1 (stationary phase measured as the elution time of toluene) and
convention 3 (stationary phase as the amount of the excess uptake of water)—
Table 2.
As the differences in the volumes of the mobile phase according to
convention 3,
Vm =
Vi +
V0 −
Vex, differ (are larger by several per cent) from the
Vm calculated according to
convention 1,
Vs =
Vex, the differences in
k are relatively more significant for the columns with larger proportions of the adsorbed water. The differences between the retention factors calculated by the two conventions depend on the composition of the mobile phase and are most significant for the zwitterionic and TSK gel columns. The differences are illustrated by several examples of the retention factors of polar phenolic compounds on the Lichrospher 100 Diol (
εH2O = 0.08) and Luna HILIC (
εH2O = 0.04) columns in 98% acetonitrile/water mobile phase in
Table 3. As can be expected, the relative differences in the retention factors are larger (approximately + 12%) for the Diol column showing approximately twice higher adsorbed water amount in comparison to the Luna HILIC column (approximately + 5%). The examples in the table show that the adopted stationary phase convention does not affect the predicted separation selectivity of sample components (the relative retention, i.e., the ratio of the retention factors,
kj/ki)—phenolic acids and flavones.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}