
Enhancement of Anticancer Potential of Pterostilbene Derivative by Chalcone Hybridization

 ,
,
Abstract
:1. Introduction
2. Chemistry
3. Biological Results and Discussion
3.1. Screening of Compound 2 and 4 Series by Cytotoxic Activity
3.2. Structure–Activity Relationship
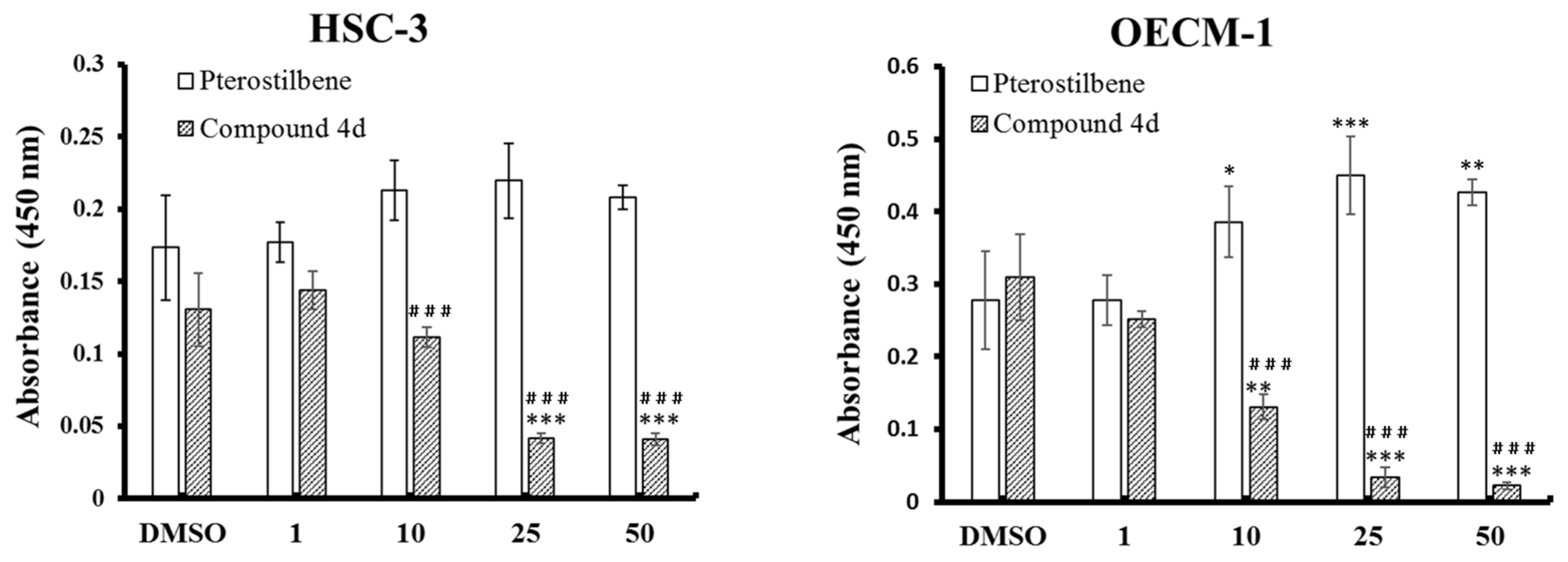
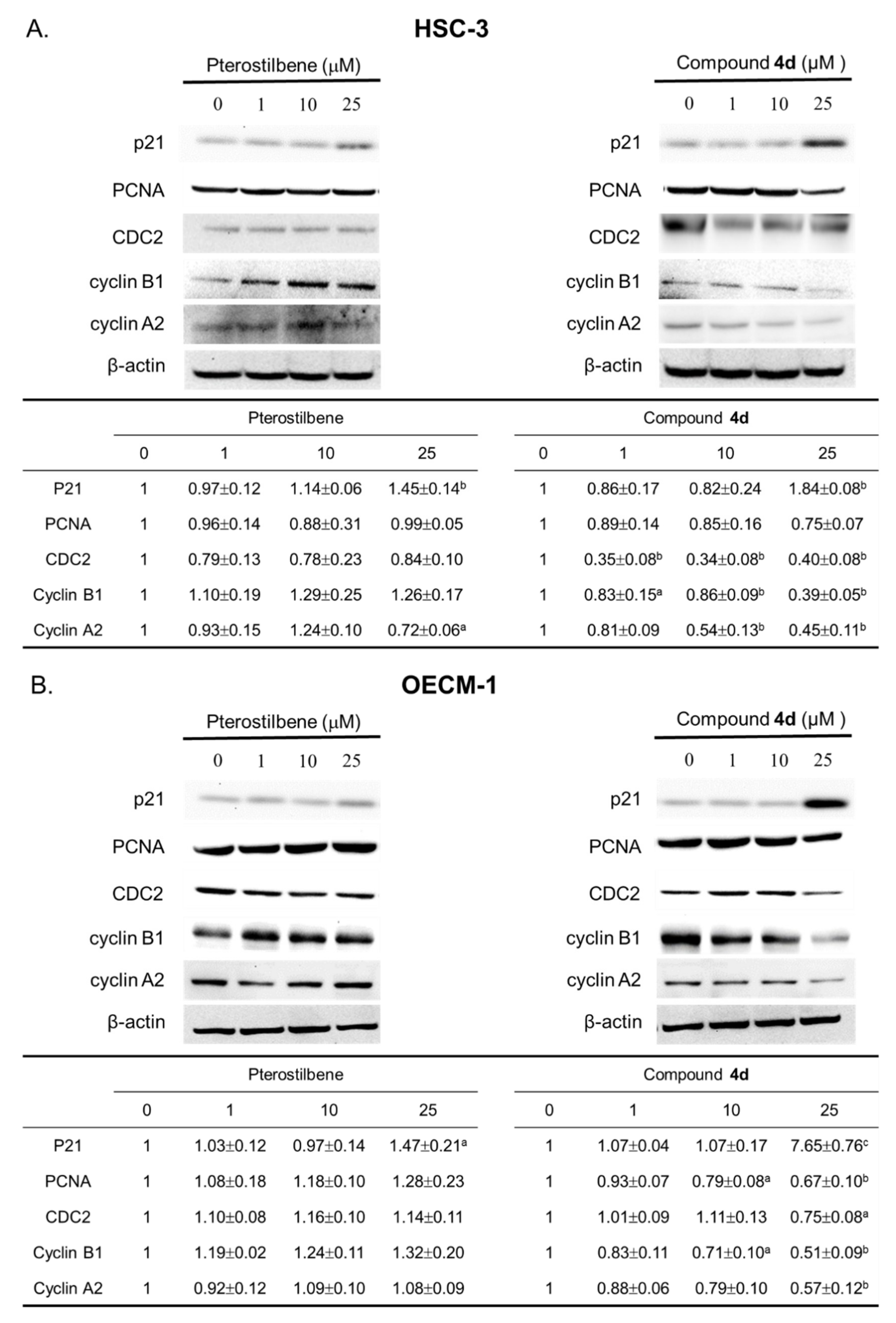
3.3. Compound 4d Effectively Inhibited the Cell Proliferation through Modulation of Cell Cycle-Regulatory Proteins
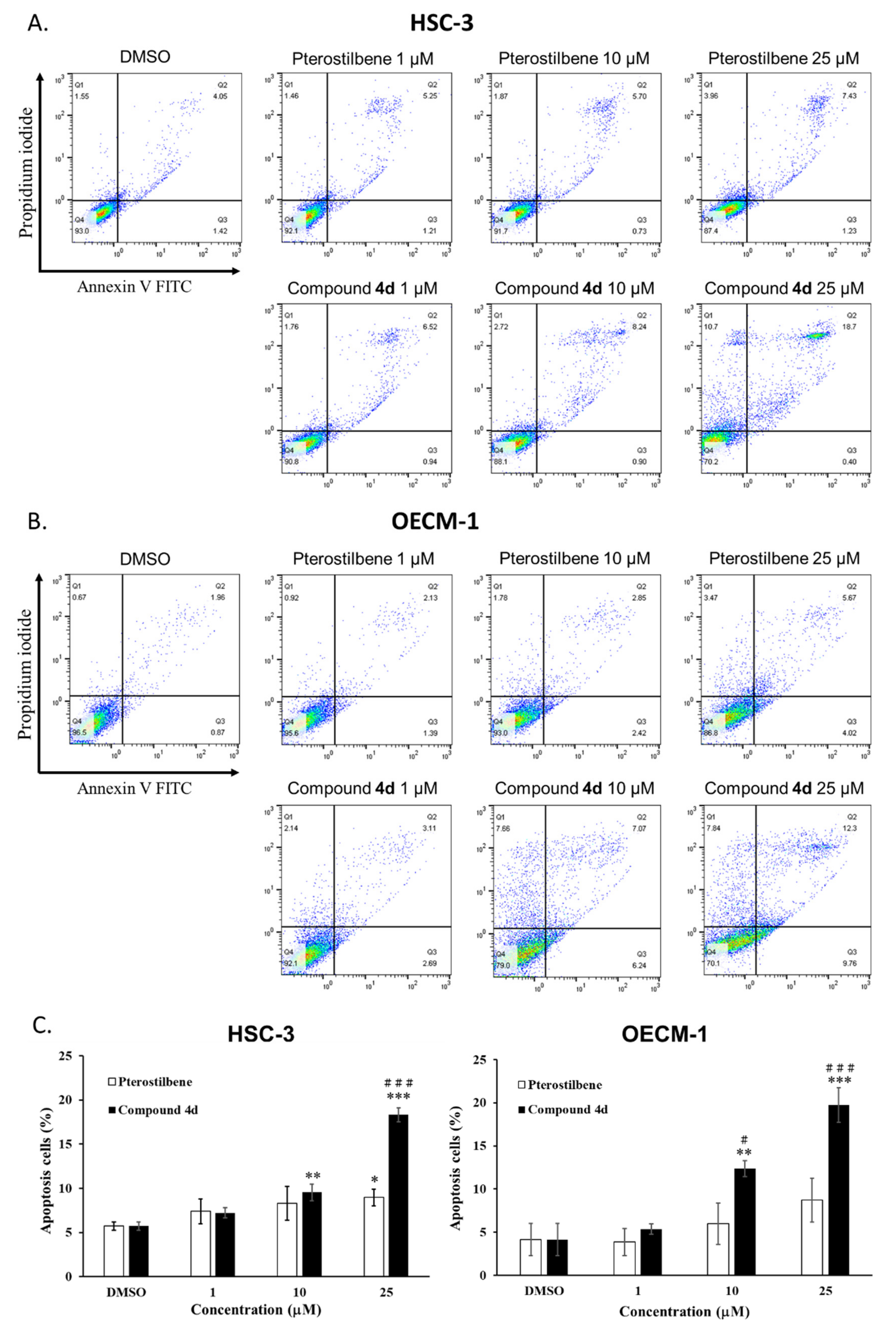
3.4. Compound 4d Induced Apoptosis through Regulation of PARP and Caspase Activation
4. Materials and Methods
4.1. General Information
4.2. General Procedure for the Synthesis of Compound 2a–i
4.2.1. 4-((E)-3,5-Dimethoxystyryl)phenyl Cinnamate (2a)
4.2.2. 4-((E)-3,5-Dimethoxystyryl)phenyl (E)-3-(4-methoxyphenyl)acrylate (2b)
4.2.3. 4-((E)-3,5-Dimethoxystyryl)phenyl (E)-3-(3-methoxyphenyl)acrylate (2c)
4.2.4. 4-((E)-3,5-Dimethoxystyryl)phenyl (E)-3-(2-methoxyphenyl)acrylate (2d)
4.2.5. 4-((E)-3,5-Dimethoxystyryl)phenyl (E)-3-(4-fluorophenyl)acrylate (2e)
4.2.6. 4-((E)-3,5-Dimethoxystyryl)phenyl (E)-3-(3-fluorophenyl)acrylate (2f)
4.2.7. 4-((E)-3,5-Dimethoxystyryl)phenyl (E)-3-(2-fluorophenyl)acrylate (2g)
4.2.8. 4-((E)-3,5-Dimethoxystyryl)phenyl (E)-3-(4-hydroxyphenyl)acrylate (2h)
4.2.9. 4-((E)-3,5-Dimethoxystyryl)phenyl (E)-3-(3-hydroxyphenyl)acrylate (2i)
4.3. Procedure for the Synthesis of 4-((E)-3,5-Dimethoxystyryl)phenyl (E)-3-(3,4-dihydroxyphenyl) Acrylate (2j)
4.4. Procedure for the Synthesis of (E)-2-(4-Hydroxystyryl)-4,6-dimethoxybenzaldehyde (3)
4.5. General Procedure for the Synthesis of Compound 4a–i
4.5.1. (E)-3-(2-((E)-4-Hydroxystyryl)-4,6-dimethoxyphenyl)-1-phenylprop-2-en-1-one (4a)
4.5.2. (E)-3-(2-((E)-4-Hydroxystyryl)-4,6-dimethoxyphenyl)-1-(4-methoxyphenyl)prop-2-en-1-one (4b)
4.5.3. (E)-3-(2-((E)-4-Hydroxystyryl)-4,6-dimethoxyphenyl)-1-(3-methoxyphenyl)prop-2-en-1-one (4c)
4.5.4. (E)-3-(2-((E)-4-Hydroxystyryl)-4,6-dimethoxyphenyl)-1-(2-methoxyphenyl)prop-2-en-1-one (4d)
4.5.5. (E)-1-(4-Fluorophenyl)-3-(2-((E)-4-hydroxystyryl)-4,6-dimethoxyphenyl)prop-2-en-1-one (4e)
4.5.6. (E)-1-(3-Fluorophenyl)-3-(2-((E)-4-hydroxystyryl)-4,6-dimethoxyphenyl)prop-2-en-1-one (4f)
4.5.7. (E)-1-(2-Fluorophenyl)-3-(2-((E)-4-hydroxystyryl)-4,6-dimethoxyphenyl)prop-2-en-1-one (4g)
4.5.8. (E)-1-(4-Hydroxyphenyl)-3-(2-((E)-4-hydroxystyryl)-4,6-dimethoxyphenyl)prop-2-en-1-one (4h)
4.5.9. (E)-1-(3-Hydroxyphenyl)-3-(2-((E)-4-hydroxystyryl)-4,6-dimethoxyphenyl)prop-2-en-1-one (4i)
4.6. Biological Studies
4.6.1. Cell Viability Assays
4.6.2. BrdU Incorporation Assay
4.6.3. Apoptosis Detection by Annexin V Staining and Flow Cytometry
4.6.4. Cell Cycle Analysis by Flow Cytometry
4.6.5. Western Blot Assay
4.6.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ma, Z.; Zhang, X.; Xu, L.; Liu, D.; Di, S.; Li, W.; Jiao, Z.; Hongmei, Z.; Xiaofei, L.; Jing, H.; et al. Pterostilbene: Mechanisms of its action as oncostatic agent in cell models and in vivo studies. Pharmacol. Res. 2019, 145, 104265. [Google Scholar] [CrossRef]
- McCormack, D.; McFadden, D. A review of pterostilbene antioxidant activity and disease modification. Oxid. Med. Cell. Longev. 2013, 2013, 575482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.C.; Tseng, C.H.; Wang, P.W.; Lu, P.L.; Weng, Y.H.; Yen, F.L.; Fang, J.Y. Pterostilbene, a Methoxylated Resveratrol Derivative, Efficiently Eradicates Planktonic, Biofilm, and Intracellular MRSA by Topical Application. Front. Microbiol. 2017, 8, 1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, M.H.; Chiou, Y.S.; Chen, W.J.; Wang, J.M.; Badmaev, V.; Ho, C.T. Pterostilbene inhibited tumor invasion via suppressing multiple signal transduction pathways in human hepatocellular carcinoma cells. Carcinogenesis 2009, 30, 1234–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, M.H.; Chang, Y.H.; Badmaev, V.; Nagabhushanam, K.; Ho, C.T. Pterostilbene induces apoptosis and cell cycle arrest in human gastric carcinoma cells. J. Agric. Food Chem. 2007, 55, 7777–7785. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Bodipati, N.; Demonacos, M.K.; Peddinti, R.; Ghosh, K.; Roy, P. Long term induction by pterostilbene results in autophagy and cellular differentiation in MCF-7 cells via ROS dependent pathway. Mol. Cell. Endocrinol. 2012, 355, 25–40. [Google Scholar] [CrossRef]
- Pan, M.H.; Lai, C.S.; Wu, J.C.; Ho, C.T. Molecular mechanisms for chemoprevention of colorectal cancer by natural dietary compounds. Mol. Nutr. Food Res. 2011, 55, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Riche, D.M.; McEwen, C.L.; Riche, K.D.; Sherman, J.J.; Wofford, M.R.; Deschamp, D.; Griswold, M. Analysis of safety from a human clinical trial with pterostilbene. J. Toxicol. 2013, 2013, 463595. [Google Scholar] [CrossRef]
- Dhar, S.; Kumar, A.; Rimando, A.M.; Zhang, X.; Levenson, A.S. Resveratrol and pterostilbene epigenetically restore PTEN expression by targeting oncomiRs of the miR-17 family in prostate cancer. Oncotarget 2015, 6, 27214–27226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, S.; Kumar, A.; Zhang, L.; Rimando, A.M.; Lage, J.M.; Lewin, J.R.; Atfi, A.; Zhang, X.; Levenson, A.S. Dietary pterostilbene is a novel MTA1-targeted chemopreventive and therapeutic agent in prostate cancer. Oncotarget 2016, 7, 18469–18484. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Yang, Y.; Fan, C.; Di, S.; Hu, W.; Jiang, S.; Li, T.; Ma, Z.; Chao, D.; Feng, X.; et al. Pterostilbene Inhibits the Growth of Human Esophageal Cancer Cells by Regulating Endoplasmic Reticulum Stress. Cell Physiol. Biochem. 2016, 38, 1226–1244. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Liu, Y.; Sharma, S.; Wu, K.; Chan, M.D.; Lo, H.W.; Carpenter, R.L.; Metheny-Barlow, L.J.; Zhou, X.; Qasem, S.A.; et al. Activation of the c-Met Pathway Mobilizes an Inflammatory Network in the Brain Microenvironment to Promote Brain Metastasis of Breast Cancer. Cancer Res. 2016, 76, 4970–4980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.J.; Han, J.M.; Choi, Y.S.; Jung, H. Pterostilbene Suppresses both Cancer Cells and Cancer Stem-Like Cells in Cervical Cancer with Superior Bioavailability to Resveratrol. Molecules 2020, 25, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.C.; Lai, C.S.; Chung, M.C.; Kalyanam, N.; Majeed, M.; Ho, C.T.; Ho, Y.-S.; Pan, M.-H. Potent anti-cancer effect of 3′-hydroxypterostilbene in human colon xenograft tumors. PLoS ONE 2014, 9, e111814. [Google Scholar] [CrossRef] [Green Version]
- Nikhil, K.; Sharan, S.; Chakraborty, A.; Bodipati, N.; Peddinti, R.K.; Roy, P. Role of isothiocyanate conjugate of pterostilbene on the inhibition of MCF-7 cell proliferation and tumor growth in Ehrlich ascitic cell induced tumor bearing mice. Exp. Cell Res. 2014, 320, 311–328. [Google Scholar] [CrossRef]
- Hsieh, M.T.; Chen, H.P.; Lu, C.C.; Chiang, J.H.; Wu, T.S.; Kuo, D.H.; Huang, L.-J.; Kuo, S.-C.; Yang, J.-S. The novel pterostilbene derivative ANK-199 induces autophagic cell death through regulating PI3 kinase class III/beclin 1/Atgrelated proteins in cisplatin-resistant CAR human oral cancer cells. Int. J. Oncol. 2014, 45, 782–794. [Google Scholar] [CrossRef]
- Tang, K.W.; Yang, S.C.; Tseng, C.H. Design, Synthesis, and Anti-Bacterial Evaluation of Triazolyl-Pterostilbene Derivatives. Int. J. Mol. Sci. 2019, 20, 4564. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, M.T.; Huang, L.J.; Wu, T.S.; Lin, H.Y.; Morris-Natschke, S.L.; Lee, K.H.; Kuo, S.C. Synthesis and antitumor activity of bis(hydroxymethyl)propionate analogs of pterostilbene in cisplatin-resistant human oral cancer cells. Bioorg. Med. Chem. 2018, 26, 3909–3916. [Google Scholar] [CrossRef]
- Kantevari, S.; Addla, D.; Bagul, P.K.; Sridhar, B.; Banerjee, S.K. Synthesis and evaluation of novel 2-butyl-4-chloro-1-methylimidazole embedded chalcones and pyrazoles as angiotensin converting enzyme (ACE) inhibitors. Bioorg. Med. Chem. 2011, 19, 4772–4781. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Asati, V.; Bharti, S.K. Chalcones and their therapeutic targets for the management of diabetes: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2015, 92, 839–865. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lim, S.S.; Shin, K.H.; Kim, Y.S.; Ohuchi, K.; Jung, S.H. Anti-angiogenic and anti-tumor activities of 2′-hydroxy-4′-methoxychalcone. Biol. Pharm. Bull. 2006, 29, 1028–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Anti-cancer chalcones: Structural and molecular target perspectives. Eur. J. Med. Chem. 2015, 98, 69–114. [Google Scholar] [CrossRef]
- Rizvi, S.U.F.; Siddiqui, H.L.; Johns, M.; Detorio, M.; Schinazi, R.F. Anti-HIV-1 and cytotoxicity studies of piperidyl-thienyl chalcones and their 2-pyrazoline derivatives. Med. Chem. Res. 2012, 21, 3741–3749. [Google Scholar] [CrossRef]
- Israf, D.A.; Khaizurin, T.A.; Syahida, A.; Lajis, N.H.; Khozirah, S. Cardamonin inhibits COX and iNOS expression via inhibition of p65NF-kappaB nuclear translocation and Ikappa-B phosphorylation in RAW 264.7 macrophage cells. Mol. Immunol. 2007, 44, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, M.I.; Mahmood, A.; Madni, M.; Masood, S.; Kashif, M. Synthesis, characterization, theoretical, anti-bacterial and molecular docking studies of quinoline based chalcones as a DNA gyrase inhibitor. Bioorg. Chem. 2014, 54, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.T.; Chen, Y.S.; Tang, K.W.; Lee, J.C.; Tseng, C.H.; Tzeng, C.C.; Yen, C.-H.; Chen, Y.L. Discovery of 4-Anilinoquinolinylchalcone Derivatives as Potential NRF2 Activators. Molecules 2020, 25, 3133. [Google Scholar] [CrossRef]
- Sahu, N.K.; Balbhadra, S.S.; Choudhary, J.; Kohli, D.V. Exploring pharmacological significance of chalcone scaffold: A review. Curr. Med. Chem. 2012, 19, 209–225. [Google Scholar] [CrossRef]
- Tseng, C.H.; Chen, Y.L.; Hsu, C.Y.; Chen, T.C.; Cheng, C.M.; Tso, H.C.; Lu, Y.-J.; Tzeng, C.C. Synthesis and antiproliferative evaluation of 3-phenylquinolinylchalcone derivatives against non-small cell lung cancers and breast cancers. Eur. J. Med. Chem. 2013, 59, 274–282. [Google Scholar] [CrossRef]
- Tseng, C.H.; Tzeng, C.C.; Hsu, C.Y.; Cheng, C.M.; Yang, C.N.; Chen, Y.L. Discovery of 3-phenylquinolinylchalcone derivatives as potent and selective anticancer agents against breast cancers. Eur. J. Med. Chem. 2015, 97, 306–319. [Google Scholar] [CrossRef]
- De, P.; Baltas, M.; Bedos-Belval, F. Cinnamic acid derivatives as anticancer agents-a review. Curr. Med. Chem. 2011, 18, 1672–1703. [Google Scholar] [CrossRef]
- Ruan, B.F.; Lu, X.; Tang, J.F.; Wei, Y.; Wang, X.L.; Zhang, Y.B.; Wang, L.S.; Zhu, H.L. Synthesis, biological evaluation, and molecular docking studies of resveratrol derivatives possessing chalcone moiety as potential antitubulin agents. Bioorg. Med. Chem. 2011, 19, 2688–2695. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Lee, J.; Park, J.; Lee, Y.; Ahn, S.; Lee, J.H.; Koh, D.; Lee, Y.H.; Lim, Y. Design, synthesis, and biological activities of 1-aryl-(3-(2-styryl)phenyl)prop-2-en-1-ones. Bioorg. Chem. 2019, 83, 438. [Google Scholar] [CrossRef] [PubMed]
- Waga, S.; Hannon, G.J.; Beach, D.; Stillman, B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 1994, 369, 574–578. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) of MTT Assay after 72 h | ||
|---|---|---|---|
| No. | R | OECM-1 | HSC-3 |
| 2a | H | >50 | >50 |
| 2b | 4-OMe | >50 | >50 |
| 2c | 3-OMe | >50 | >50 |
| 2d | 2-OMe | >50 | >50 |
| 2e | 4-F | >50 | >50 |
| 2f | 3-F | >50 | >50 |
| 2g | 2-F | 40.24 ± 0.06 | >50 |
| 2h | 4-OH | >50 | >50 |
| 2i | 3-OH | >50 | >50 |
| 2j | 3,4-diOH | 22.26 ± 0.24 | 18.53 ± 0.43 |
| 4a | H | 22.80 ± 0.27 | 43.44 ± 0.75 |
| 4b | 4-OMe | 11.08 ± 0.05 | 28.93 ± 0.09 |
| 4c | 3-OMe | 11.50 ± 0.01 | 18.22 ± 0.11 |
| 4d | 2-OMe | 16.38 ± 0.10 | 18.06 ± 0.05 |
| 4e | 4-F | 13.70 ± 0.01 | 19.13 ± 0.26 |
| 4f | 3-F | 16.39 ± 0.03 | 20.41 ± 0.41 |
| 4g | 2-F | 17.34 ± 0.15 | 18.43 ± 0.09 |
| 4h | 4-OH | 46.21 ± 2.01 | >50 |
| 4i | 3-OH | 9.62 ± 0.34 | 19.45 ± 0.11 |
| Pterostilbene | 40.19 ± 0.69 | >50 | |
| IC50 (µM) of Compound 4d | ||
|---|---|---|
| Time (h) | HSC-3 | OECM-1 |
| 24 | 21.72 ± 0.55 | 25.07 ± 0.67 |
| 48 | 17.98 ± 0.41 a | 20.47 ± 0.55 a |
| 72 | 18.06 ± 0.05 a | 16.38 ± 0.10 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, K.-W.; Ke, C.-C.; Tseng, C.-H.; Chen, Y.-L.; Tzeng, C.-C.; Chen, Y.-J.; Hsu, C.-C.; Tai, H.-T.; Hsieh, Y.-J. Enhancement of Anticancer Potential of Pterostilbene Derivative by Chalcone Hybridization. Molecules 2021, 26, 4840. https://doi.org/10.3390/molecules26164840
Tang K-W, Ke C-C, Tseng C-H, Chen Y-L, Tzeng C-C, Chen Y-J, Hsu C-C, Tai H-T, Hsieh Y-J. Enhancement of Anticancer Potential of Pterostilbene Derivative by Chalcone Hybridization. Molecules. 2021; 26(16):4840. https://doi.org/10.3390/molecules26164840
Chicago/Turabian StyleTang, Kai-Wei, Chien-Chih Ke, Chih-Hua Tseng, Yeh-Long Chen, Cherng-Chyi Tzeng, Yi-Jin Chen, Chia-Chi Hsu, Hsiao-Ting Tai, and Ya-Ju Hsieh. 2021. "Enhancement of Anticancer Potential of Pterostilbene Derivative by Chalcone Hybridization" Molecules 26, no. 16: 4840. https://doi.org/10.3390/molecules26164840
APA StyleTang, K. -W., Ke, C. -C., Tseng, C. -H., Chen, Y. -L., Tzeng, C. -C., Chen, Y. -J., Hsu, C. -C., Tai, H. -T., & Hsieh, Y. -J. (2021). Enhancement of Anticancer Potential of Pterostilbene Derivative by Chalcone Hybridization. Molecules, 26(16), 4840. https://doi.org/10.3390/molecules26164840