
The Nrf2 Pathway in Ischemic Stroke: A Review



 , and
, and 
Abstract
:1. Introduction
2. The Ischemic Cascade and Oxidative Consequences
3. Nrf2 Signaling Pathway and Ischemic Stroke
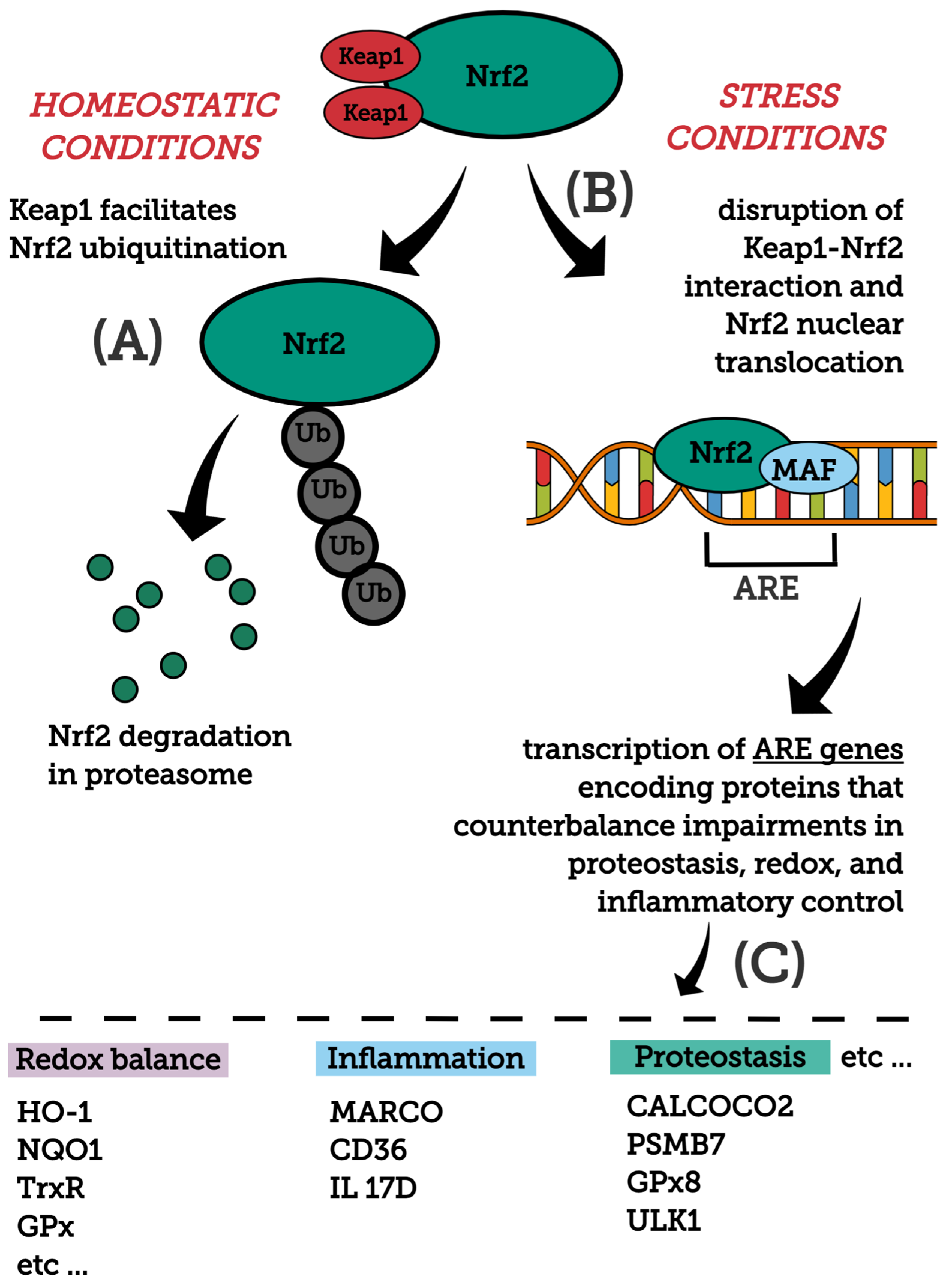
3.1. Overview of the Nrf2 Signaling Pathway
3.2. Molecular Events Linking Ischemic Stroke and Nrf2 Pathway
3.3. Effects of Nrf2 Modulators in Ischemic Stroke: Evidence from Experimental Studies
3.3.1. Curcumin
3.3.2. Fumarate
3.3.3. Resveratrol
3.3.4. Sulforaphane
3.3.5. Tert-Butylhydroquinone
3.3.6. Carbon Monoxide
4. Challenges/Perspectives on the Use of Nrf2 Activators in Ischemic Stroke Patients
Funding
Conflicts of Interest
References
- Taoufik, E.; Probert, L. Ischemic Neuronal Damage. CPD 2008, 14, 3565–3573. [Google Scholar] [CrossRef]
- Shakir, R.; Norrving, B. Stroke in ICD-11: The end of a long exile. Lancet 2017, 389, 2373. [Google Scholar] [CrossRef] [Green Version]
- George, M.G.; Tong, X.; Kuklina, E.V.; Labarthe, D.R. Trends in stroke hospitalizations and associated risk factors among children and young adults, 1995–2008. Ann. Neurol. 2011, 70, 713–721. [Google Scholar] [CrossRef]
- Appelros, P.; Stegmayr, B.; Terént, A. Sex Differences in Stroke Epidemiology: A Systematic Review. Stroke 2009, 40, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M. The sixth report of the JoInt. National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure, and 1999 World Health Organization-International Society of Hypertension Guidelines for the Management of Hypertension. Nihon Rinsho 2000, 58 (Suppl. 1), 267–275. [Google Scholar]
- Seshadri, S.; Beiser, A.; Pikula, A.; Himali, J.J.; Kelly-Hayes, M.; Debette, S.; DeStefano, A.L.; Romero, J.R.; Kase, C.S.; Wolf, P.A. Parental Occurrence of Stroke and Risk of Stroke in Their Children: The Framingham Study. Circulation 2010, 121, 1304–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, M.J.; Xavier, D.; Liu, L.; Zhang, H.; Chin, S.L.; Rao-Melacini, P.; Rangarajan, S.; Islam, S.; Pais, P.; McQueen, M.J.; et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): A case-control study. Lancet 2010, 376, 112–123. [Google Scholar] [CrossRef]
- Vermeer, S.E.; Sandee, W.; Algra, A.; Koudstaal, P.J.; Kappelle, L.J.; Dippel, D.W.J. Impaired Glucose Tolerance Increases Stroke Risk in Nondiabetic Patients With Transient Ischemic Attack or Minor Ischemic Stroke. Stroke 2006, 37, 1413–1417. [Google Scholar] [CrossRef] [Green Version]
- Romero, J.R.; Morris, J.; Pikula, A. Stroke prevention: Modifying risk factors. Ther. Adv. Cardiovasc. Dis. 2008, 2, 287–303. [Google Scholar] [CrossRef] [Green Version]
- Denti, L.; Cecchetti, A.; Annoni, V.; Merli, M.F.; Ablondi, F.; Valenti, G. The role of lipid profile in determining the risk of ischemic stroke in the elderly: A case–control study. Arch. Gerontol. Geriatr. 2003, 37, 51–62. [Google Scholar] [CrossRef]
- Bhat, V.M.; Cole, J.W.; Sorkin, J.D.; Wozniak, M.A.; Malarcher, A.M.; Giles, W.H.; Stern, B.J.; Kittner, S.J. Dose-Response Relationship Between Cigarette Smoking and Risk of Ischemic Stroke in Young Women. Stroke 2008, 39, 2439–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonnell, M.N.; Hillier, S.L.; Hooker, S.P.; Le, A.; Judd, S.E.; Howard, V.J. Physical Activity Frequency and Risk of Incident Stroke in a National US Study of Blacks and Whites. Stroke 2013, 44, 2519–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estruch, R.; Ros, E.; Martínez-González, M.A. Mediterranean Diet for Primary Prevention of Cardiovascular Disease. N. Engl. J. Med. 2013, 369, 672–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuriakose, D.; Xiao, Z. Pathophysiology and Treatment of Stroke: Present Status and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 7609. [Google Scholar] [CrossRef] [PubMed]
- Hacke, W.; Kaste, M.; Bluhmki, E.; Brozman, M.; Dávalos, A.; Guidetti, D.; Larrue, V.; Lees, K.R.; Medeghri, Z.; Machnig, T.; et al. Thrombolysis with Alteplase 3 to 4.5 Hours after Acute Ischemic Stroke. N. Engl. J. Med. 2008, 359, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Rabinstein, A.A. Update on Treatment of Acute Ischemic Stroke. Continuum 2020, 26, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Orellana-Urzúa, S.; Rojas, I.; Líbano, L.; Rodrigo, R. Pathophysiology of Ischemic Stroke: Role of Oxidative Stress. CPD 2020, 26, 4246–4260. [Google Scholar] [CrossRef]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef]
- Kaplan, J.H. Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 2002, 71, 511–535. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Tymianski, M. Molecular mechanisms of calcium-dependent excitotoxicity. J. Mol. Med. 2000, 78, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Depp, C.; Bas-Orth, C.; Schroeder, L.; Hellwig, A.; Bading, H. Synaptic Activity Protects Neurons Against Calcium-Mediated Oxidation and Contraction of Mitochondria During Excitotoxicity. Antioxid. Redox Signal. 2018, 29, 1109–1124. [Google Scholar] [CrossRef] [PubMed]
- Quillinan, N.; Herson, P.S.; Traystman, R.J. Neuropathophysiology of Brain Injury. Anesthesiol. Clin. 2016, 34, 453–464. [Google Scholar] [CrossRef] [Green Version]
- Saito, A.; Maier, C.M.; Narasimhan, P.; Nishi, T.; Song, Y.S.; Yu, F.; Liu, J.; Lee, Y.-S.; Nito, C.; Kamada, H.; et al. Oxidative Stress and Neuronal Death/Survival Signaling in Cerebral Ischemia. Mol. Neurobiol. 2005, 31, 105–116. [Google Scholar] [CrossRef]
- Epstein, F.H.; McCord, J.M. Oxygen-Derived Free Radicals in Postischemic Tissue Injury. N. Engl. J. Med. 1985, 312, 159–163. [Google Scholar] [CrossRef]
- Uyama, O.; Matsuyama, T.; Michishita, H.; Nakamura, H.; Sugita, M. Protective effects of human recombinant superoxide dismutase on transient ischemic injury of CA1 neurons in gerbils. Stroke 1992, 23, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Morooka, H.; Hirotsune, N.; Wani, T.; Ohmoto, T. Histochemical Demonstration of Free Radicals (H22O2) in Ischemic Brain Edema and Protective Effects of Human Recombinant Superoxide Dismutase on Ischemic Neuronal Damage. In Brain Edema IX; Ito, U., Baethmann, A., Hossmann, K.-A., Kuroiwa, T., Marmarou, A., Reulen, H.-J., Takakura, K., Eds.; Springer: Vienna, Austria, 1994; pp. 307–309. ISBN 978-3-7091-9336-5. [Google Scholar]
- Kinuta, Y.; Kikuchi, H.; Ishikawa, M. Ischaemic Brain Oedema and Xanthine-Xanthine Oxidase System. In Brain Edema VIII; Reulen, H.-J., Baethmann, A., Fenstermacher, J., Marmarou, A., Spatz, M., Eds.; Springer: Vienna, Austria, 1990; pp. 192–194. ISBN 978-3-7091-9117-0. [Google Scholar]
- Muralikrishna Adibhatla, R.; Hatcher, J.F. Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic. Biol. Med. 2006, 40, 376–387. [Google Scholar] [CrossRef]
- Nakahara, I.; Kikuchi, H.; Taki, W.; Nishi, S.; Kito, M.; Yonekawa, Y.; Goto, Y.; Ogata, N. Changes in major phospholipids of mitochondria during postischemic reperfusion in rat brain. J. Neurosurg. 1992, 76, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Chomova, M.; Zitnanova, I. Look into brain energy crisis and membrane pathophysiology in ischemia and reperfusion. Stress 2016, 19, 341–348. [Google Scholar] [CrossRef]
- Lake, E.M.R.; Mester, J.; Thomason, L.A.; Adams, C.; Bazzigaluppi, P.; Koletar, M.; Janik, R.; Carlen, P.; McLaurin, J.; Stanisz, G.J.; et al. Modulation of the peri-infarct neurogliovascular function by delayed COX-1 inhibition: Delayed COX-1 Inhibition Stroke Treatment. J. Magn. Reson. Imaging 2017, 46, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Yang, Y.; DeMars, K.M.; Rosenberg, G.A.; Candelario-Jalil, E. Genetic Deletion or Pharmacological Inhibition of Cyclooxygenase-2 Reduces Blood-Brain Barrier Damage in Experimental Ischemic Stroke. Front. Neurol. 2020, 11, 887. [Google Scholar] [CrossRef] [PubMed]
- Leyen, K.V. Lipoxygenase: An Emerging Target for Stroke Therapy. CNSNDDT 2013, 12, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zuo, F.; Wu, H. Blockage of cytosolic phospholipase A2 alpha by monoclonal antibody attenuates focal ischemic brain damage in mice. Biosci Trends 2017, 11, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Yu, Y. Small molecules targeting cyclooxygenase/prostanoid cascade in experimental brain ischemia: Do they translate? Med. Res. Rev. 2021, 41, 828–857. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Ago, T.; Kitazono, T.; Nabika, T. NADPH Oxidase-Related Pathophysiology in Experimental Models of Stroke. Int. J. Mol. Sci. 2017, 18, 2123. [Google Scholar] [CrossRef] [Green Version]
- Casas, A.I.; Kleikers, P.W.M.; Geuss, E.; Langhauser, F.; Adler, T.; Busch, D.H.; Gailus-Durner, V.; de Angelis, M.H.; Egea, J.; Lopez, M.G.; et al. Calcium-dependent blood-brain barrier breakdown by NOX5 limits postreperfusion benefit in stroke. J. Clin. Investig. 2019, 129, 1772–1778. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, Y.; Zhao, X.; Shao, L.; Liu, G.; Sun, C.; Xu, R.; Zhang, Z. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav. Immun. 2021, 93, 312–321. [Google Scholar] [CrossRef]
- Kimura-Ohba, S.; Yang, Y. Oxidative DNA Damage Mediated by Intranuclear MMP Activity Is Associated with Neuronal Apoptosis in Ischemic Stroke. Oxid. Med. Cell. Longev. 2016, 2016, 6927328. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, H.; Wang, L.; Konishi, T. Prevention of Cerebral Oxidative Injury by Post-ischemic Intravenous Administration of Shengmai San. Am. J. Chin. Med. 2006, 34, 591–600. [Google Scholar] [CrossRef]
- Narne, P.; Pandey, V.; Phanithi, P.B. Role of Nitric Oxide and Hydrogen Sulfide in Ischemic Stroke and the Emergent Epigenetic Underpinnings. Mol. Neurobiol. 2019, 56, 1749–1769. [Google Scholar] [CrossRef]
- Dogan, O.; Kisa, U.; Erdemoglu, A.K.; Kacmaz, M.; Caglayan, O.; Kurku, H. Oxidative and nitrosative stress in patients with ischemic stroke. LaboratoriumsMedizin 2018, 42, 195–200. [Google Scholar] [CrossRef]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, M.A.; Hayes, J.D. The Keap1/Nrf2 pathway in health and disease: From the bench to the clinic. Biochem. Soc. Trans. 2015, 43, 687–689. [Google Scholar] [CrossRef] [Green Version]
- Sova, M.; Saso, L. Design and development of Nrf2 modulators for cancer chemoprevention and therapy: A review. Drug Des. Dev. Ther. 2018, 12, 3181–3197. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Gonçalves, F.M.; Abiko, Y.; Li, H.; Kumagai, Y.; Aschner, M. Redox toxicology of environmental chemicals causing oxidative stress. Redox Biol. 2020, 34, 101475. [Google Scholar] [CrossRef]
- Cores, Á.; Piquero, M.; Villacampa, M.; León, R.; Menéndez, J.C. NRF2 Regulation Processes as a Source of Potential Drug Targets against Neurodegenerative Diseases. Biomolecules 2020, 10, 904. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP Augments Promoter-Specific DNA Binding of Nrf2 during the Antioxidant Response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [Green Version]
- Theodore, M.; Kawai, Y.; Yang, J.; Kleshchenko, Y.; Reddy, S.P.; Villalta, F.; Arinze, I.J. Multiple Nuclear Localization Signals Function in the Nuclear Import of the Transcription Factor Nrf2. J. Biol. Chem. 2008, 283, 8984–8994. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [Green Version]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The Carboxy-Terminal Neh3 Domain of Nrf2 Is Required for Transcriptional Activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [Green Version]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription: Synergistic activation of Nrf2 by CBP. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef]
- Krajka-Kuźniak, V.; Paluszczak, J.; Baer-Dubowska, W. The Nrf2-ARE signaling pathway: An update on its regulation and possible role in cancer prevention and treatment. Pharmacol. Rep. 2017, 69, 393–402. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobón-Velasco, J.C.; Devijver, H.; García-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/β-TrCP axis. Mol. Cell. Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRα Inhibits the NRF2-ARE Signaling Pathway through a Direct Interaction with the Neh7 Domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Mittal, R. Nrf2: A potential therapeutic target for diabetic neuropathy. Inflammopharmacology 2017, 25, 393–402. [Google Scholar] [CrossRef] [PubMed]
- David, J.A.; Rifkin, W.J.; Rabbani, P.S.; Ceradini, D.J. The Nrf2/Keap1/ARE Pathway and Oxidative Stress as a Therapeutic Target in Type II Diabetes Mellitus. J. Diabetes Res. 2017, 2017, 4826724. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB Protein Is an Adaptor That Bridges Nrf2 to a Cul3-Based E3 Ligase: Oxidative Stress Sensing by a Cul3-Keap1 Ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Lo, S.-C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 Is a Redox-Regulated Substrate Adaptor Protein for a Cul3-Dependent Ubiquitin Ligase Complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer Chemoprevention Mechanisms Mediated through the Keap1–Nrf2 Pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- Sykiotis, G.P.; Bohmann, D. Stress-Activated Cap’n’collar Transcription Factors in Aging and Human Disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, S.; Pal, D.; Sandur, S.K. Nrf2 facilitates repair of radiation induced DNA damage through homologous recombination repair pathway in a ROS independent manner in cancer cells. Mutat. Res. 2015, 779, 33–45. [Google Scholar] [CrossRef]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef] [Green Version]
- Niture, S.K.; Jaiswal, A.K. Nrf2-induced antiapoptotic Bcl-xL protein enhances cell survival and drug resistance. Free Radic. Biol. Med. 2013, 57, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, Y.-Z.; Miao, H.; Cheng, J.-J.; Qi, J.-M. Effects of Amelioration of Total Flavonoids from Stems and Leaves of Scutellaria baicalensis Georgi on Cognitive Deficits, Neuronal Damage and Free Radicals Disorder Induced by Cerebral Ischemia in Rats. Biol. Pharma. Bull. 2006, 29, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-H.; Kuo, H.-C.; Lee, K.-F.; Tsai, T.-H. Magnolol protects neurons against ischemia injury via the downregulation of p38/MAPK, CHOP and nitrotyrosine. Toxicol. Appl. Pharmacol. 2014, 279, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-J.; Xie, G.-N.; Liu, L.; Fu, Z.-J.; Zhang, Z.-W.; Teng, L.-Z. Sesamol attenuates oxidative stress, apoptosis and inflammation in focal cerebral ischemia/reperfusion injury. Exp. Ther. Med. 2017, 14, 841–847. [Google Scholar] [CrossRef]
- Ahmari, M.; Sharafi, A.; Mahmoudi, J.; Jafari-Anarkoli, I.; Gharbavi, M.; Hosseini, M.-J. Selegiline (l-Deprenyl) Mitigated Oxidative Stress, Cognitive Abnormalities, and Histopathological Change in Rats: Alternative Therapy in Transient Global Ischemia. J. Mol. Neurosci. 2020, 70, 1639–1648. [Google Scholar] [CrossRef]
- Liu, L.; Locascio, L.M.; Doré, S. Critical Role of Nrf2 in Experimental Ischemic Stroke. Front. Pharmacol. 2019, 10, 153. [Google Scholar] [CrossRef] [Green Version]
- Purdom-Dickinson, S.E.; Sheveleva, E.V.; Sun, H.; Chen, Q.M. Translational Control of Nrf2 Protein in Activation of Antioxidant Response by Oxidants. Mol. Pharmacol. 2007, 72, 1074–1081. [Google Scholar] [CrossRef]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [Green Version]
- Takagi, T.; Kitashoji, A.; Iwawaki, T.; Tsuruma, K.; Shimazawa, M.; Yoshimura, S.; Iwama, T.; Hara, H. Temporal activation of Nrf2 in the penumbra and Nrf2 activator-mediated neuroprotection in ischemia-reperfusion injury. Free Radic. Biol. Med. 2014, 72, 124–133. [Google Scholar] [CrossRef]
- Srivastava, S.; Alfieri, A.; Siow, R.C.M.; Mann, G.E.; Fraser, P.A. Temporal and spatial distribution of Nrf2 in rat brain following stroke: Quantification of nuclear to cytoplasmic Nrf2 content using a novel immunohistochemical technique: Quantification of cerebral Nrf2 expression in stroke. J. Physiol. 2013, 591, 3525–3538. [Google Scholar] [CrossRef]
- Dong, J.; Sulik, K.K.; Chen, S. Nrf2-Mediated Transcriptional Induction of Antioxidant Response in Mouse Embryos Exposed to Ethanol in vivo: Implications for the Prevention of Fetal Alcohol Spectrum Disorders. Antioxid. Redox Signal. 2008, 10, 2023–2033. [Google Scholar] [CrossRef] [Green Version]
- Kubo, E.; Chhunchha, B.; Singh, P.; Sasaki, H.; Singh, D.P. Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci. Rep. 2017, 7, 14130. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.-F.; Lu, J.-J.; Cao, Y.; Wang, W.; Li, H.-H.; Chen, J.-G.; Wang, F.; Wu, P.-F. Sulforaphane alleviates ethanol-mediated central inhibition and reverses chronic stress-induced aggravation of acute alcoholism via targeting Nrf2-regulated catalase expression. Neuropharmacology 2020, 176, 108235. [Google Scholar] [CrossRef]
- Dhaliwal, N.; Dhaliwal, J.; Singh, A.; Chopra, K. Dimethyl fumarate attenuates 2-VO-induced vascular dementia via activating the Nrf2 signaling pathway in rats. Inflammopharmacology 2021, 29, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y. A Small-Molecule-Inducible Nrf2-Mediated Antioxidant Response Provides Effective Prophylaxis against Cerebral Ischemia In Vivo. J. Neurosci. 2005, 25, 10321–10335. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Vollmer, M.K.; Fernandez, V.M.; Dweik, Y.; Kim, H.; Doré, S. Korean Red Ginseng Pretreatment Protects Against Long-Term Sensorimotor Deficits After Ischemic Stroke Likely Through Nrf2. Front. Cell. Neurosci. 2018, 12, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Ikeda, Y.; Ohta, Y.; Deguchi, K.; Tian, F.; Shang, J.; Matsuura, T.; Abe, K. Expression of Keap1-Nrf2 system and antioxidative proteins in mouse brain after transient middle cerebral artery occlusion. Brain Res. 2011, 1370, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Fu, B.; Zhang, X.; Zhao, T.; Chen, L.; Zhang, J.; Wang, X. Paeonol pretreatment attenuates cerebral ischemic injury via upregulating expression of pAkt, Nrf2, HO-1 and ameliorating BBB permeability in mice. Brain Res. Bull. 2014, 109, 61–67. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Cui, L.; Wang, L.; Liu, H.; Ji, H.; Du, Y. Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res. 2013, 1497, 32–39. [Google Scholar] [CrossRef]
- Nakano, Y.; Yamashita, T.; Li, Q.; Sato, K.; Ohta, Y.; Morihara, R.; Hishikawa, N.; Abe, K. Time-dependent change of in vivo optical imaging of oxidative stress in a mouse stroke model. J. Neurosci. Res. 2017, 95, 2030–2039. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Jing, X.; Wei, X.; Perez, R.G.; Ren, M.; Zhang, X.; Lou, H. S-allyl cysteine activates the Nrf2-dependent antioxidant response and protects neurons against ischemic injury in vitro and in vivo. J. Neurochem. 2015, 133, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Ya, B.-L.; Li, H.-F.; Wang, H.-Y.; Wu, F.; Xin, Q.; Cheng, H.-J.; Li, W.-J.; Lin, N.; Ba, Z.-H.; Zhang, R.-J.; et al. 5-HMF attenuates striatum oxidative damage via Nrf2/ARE signaling pathway following transient global cerebral ischemia. Cell Stress Chaperones 2017, 22, 55–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wei, R.; Zhang, L.; Tan, Y.; Qian, C. Sirtuin 6 protects the brain from cerebral ischemia/reperfusion injury through NRF2 activation. Neuroscience 2017, 366, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Yan, H.; Jiao, Y.; Ohta, Y.; Liu, X.; Li, X.; Morihara, R.; Nakano, Y.; Fukui, Y.; Shi, X.; et al. Therapeutic Effects of Pretreatment with Tocovid on Oxidative Stress in Postischemic Mice Brain. J. Stroke Cerebrovasc. Dis. 2018, 27, 2096–2105. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Lyu, Z.; Yang, X.; Pan, Z.; Lou, J.; Dong, T. Epigallocatechin-3-gallate promotes angiogenesis via up-regulation of Nfr2 signaling pathway in a mouse model of ischemic stroke. Behav. Brain Res. 2017, 321, 79–86. [Google Scholar] [CrossRef]
- Yamauchi, K.; Nakano, Y.; Imai, T.; Takagi, T.; Tsuruma, K.; Shimazawa, M.; Iwama, T.; Hara, H. A novel nuclear factor erythroid 2-related factor 2 (Nrf2) activator RS9 attenuates brain injury after ischemia reperfusion in mice. Neuroscience 2016, 333, 302–310. [Google Scholar] [CrossRef]
- Cui, H.-Y.; Zhang, X.-J.; Yang, Y.; Zhang, C.; Zhu, C.-H.; Miao, J.-Y.; Chen, R. Rosmarinic acid elicits neuroprotection in ischemic stroke via Nrf2 and heme oxygenase 1 signaling. Neural Regen. Res. 2018, 13, 2119. [Google Scholar] [CrossRef]
- Bi, F.; Zhang, Y.; Liu, W.; Xie, K. Sinomenine activation of Nrf2 signaling prevents inflammation and cerebral injury in a mouse model of ischemic stroke. Exp. Ther. Med. 2021, 21, 647. [Google Scholar] [CrossRef]
- Wang, H.; Wei, W.; Lan, X.; Liu, N.; Li, Y.; Ma, H.; Sun, T.; Peng, X.; Zhuang, C.; Yu, J. Neuroprotective Effect of Swertiamain on Cerebral Ischemia/Reperfusion Injury by Inducing the Nrf2 Protective Pathway. ACS Chem. Neurosci. 2019, 10, 2276–2286. [Google Scholar] [CrossRef]
- Yang, M.-Y.; Yu, Q.-L.; Huang, Y.-S.; Yang, G. Neuroprotective effects of andrographolide derivative CX-10 in transient focal ischemia in rat: Involvement of Nrf2/AE and TLR/NF-κB signaling. Pharmacol. Res. 2019, 144, 227–234. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, X.; Yang, Y.; Zhang, L.; Cui, L.; Zhang, C.; Chen, R.; Xie, Y.; He, J.; He, W. Tert-butylhydroquinone enhanced angiogenesis and astrocyte activation by activating nuclear factor-E2-related factor 2/heme oxygenase-1 after focal cerebral ischemia in mice. Microvasc. Res. 2019, 126, 103891. [Google Scholar] [CrossRef] [PubMed]
- Jianrong, S.; Yanjun, Z.; Chen, Y.; Jianwen, X. DUSP14 rescues cerebral ischemia/reperfusion (IR) injury by reducing inflammation and apoptosis via the activation of Nrf-2. Biochem. Biophys. Res. Commun. 2019, 509, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, W.; Lv, C.; Wang, Y.; Ma, B.; Zhang, H.; Fan, Z.; Li, M.; Li, X. A novel biscoumarin compound ameliorates cerebral ischemia reperfusion-induced mitochondrial oxidative injury via Nrf2/Keap1/ARE signaling. Neuropharmacology 2020, 167, 107918. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Fan, C.; Chen, N.; Huang, J.; Yang, Q. Resveratrol pretreatment attenuates cerebral ischemic injury by upregulating expression of transcription factor Nrf2 and HO-1 in rats. Neurochem. Res. 2011, 36, 2352–2362. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-Y.; Kuan, Y.-H.; Li, J.-R.; Chen, W.-Y.; Ou, Y.-C.; Pan, H.-C.; Liao, S.-L.; Raung, S.-L.; Chang, C.-J.; Chen, C.-J. Docosahexaenoic acid reduces cellular inflammatory response following permanent focal cerebral ischemia in rats. J. Nutr. Biochem. 2013, 24, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.-K.; Chang, C.-Y.; Ou, Y.-C.; Chen, W.-Y.; Kuan, Y.-H.; Pan, H.-C.; Liao, S.-L.; Li, G.-Z.; Chen, C.-J. Tetramethylpyrazine reduces cellular inflammatory response following permanent focal cerebral ischemia in rats. Exp. Neurol. 2013, 247, 188–201. [Google Scholar] [CrossRef]
- Alfieri, A.; Srivastava, S.; Siow, R.C.M.; Cash, D.; Modo, M.; Duchen, M.R.; Fraser, P.A.; Williams, S.C.R.; Mann, G.E. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood–brain barrier disruption and neurological deficits in stroke. Free Radic. Biol. Med. 2013, 65, 1012–1022. [Google Scholar] [CrossRef]
- Wu, J.; Li, Q.; Wang, X.; Yu, S.; Li, L.; Wu, X.; Chen, Y.; Zhao, J.; Zhao, Y. Neuroprotection by Curcumin in Ischemic Brain Injury Involves the Akt/Nrf2 Pathway. PLoS ONE 2013, 8, e59843. [Google Scholar] [CrossRef] [Green Version]
- Peng, B.; Zhao, P.; Lu, Y.-P.; Chen, M.-M.; Sun, H.; Wu, X.-M.; Zhu, L. Z-ligustilide activates the Nrf2/HO-1 pathway and protects against cerebral ischemia-reperfusion injury in vivo and in vitro. Brain Res. 2013, 1520, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Wang, M.; Jing, X.; Shi, H.; Ren, M.; Lou, H. (−)-Epigallocatechin Gallate Protects Against Cerebral Ischemia-Induced Oxidative Stress via Nrf2/ARE Signaling. Neurochem. Res. 2014, 39, 1292–1299. [Google Scholar] [CrossRef]
- Zhang, J.; Fu, B.; Zhang, X.; Zhang, L.; Bai, X.; Zhao, X.; Chen, L.; Cui, L.; Zhu, C.; Wang, L.; et al. Bicyclol upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. Bull. 2014, 100, 38–43. [Google Scholar] [CrossRef]
- Meng, H.; Guo, J.; Wang, H.; Yan, P.; Niu, X.; Zhang, J. Erythropoietin activates Keap1-Nrf2/ARE pathway in rat brain after ischemia. Int. J. Neurosci. 2014, 124, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Sun, J.; Lv, G.; Yu, Y.; Wang, G.; Xie, K.; Jiao, Y.; Yu, Y. Sevoflurane postconditioning attenuates cerebral ischemia-reperfusion injury via protein kinase B/nuclear factor-erythroid 2-related factor 2 pathway activation. Int. J. Dev. Neurosci. 2014, 38, 79–86. [Google Scholar] [CrossRef]
- Guo, H.; Li, M.; Liu, Q.; Guo, L.; Ma, M.; Wang, S.; Yu, B.; Hu, L.-M. Danhong Injection Attenuates Ischemia/Reperfusion-Induced Brain Damage Which is Associating with Nrf2 Levels In Vivo and In Vitro. Neurochem. Res. 2014, 39, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Ashabi, G.; Khalaj, L.; Khodagholi, F.; Goudarzvand, M.; Sarkaki, A. Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab. Brain Dis. 2015, 30, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-Y.; Kao, T.-K.; Chen, W.-Y.; Ou, Y.-C.; Li, J.-R.; Liao, S.-L.; Raung, S.-L.; Chen, C.-J. Tetramethylpyrazine inhibits neutrophil activation following permanent cerebral ischemia in rats. Biochem. Biophys. Res. Commun. 2015, 463, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhu, K.; Liu, Y.; Wu, X.; Wu, J.; Zhao, Y.; Zhao, J. Targeting thioredoxin-1 with siRNA exacerbates oxidative stress injury after cerebral ischemia/reperfusion in rats. Neuroscience 2015, 284, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qiu, J.; Wang, Z.; You, W.; Wu, L.; Ji, C.; Chen, G. Dimethylfumarate alleviates early brain injury and secondary cognitive deficits after experimental subarachnoid hemorrhage via activation of Keap1-Nrf2-ARE system. J. Neurosurg. 2015, 123, 915–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Park, Y.H.; Jeon, Y.T.; Hwang, J.W.; Lim, Y.J.; Kim, E.; Park, S.Y.; Park, H.P. Sevoflurane post-conditioning increases nuclear factor erythroid 2-related factor and haemoxygenase-1 expression via protein kinase C pathway in a rat model of transient global cerebral ischaemia. Br. J. Anaesth. 2015, 114, 307–318. [Google Scholar] [CrossRef] [Green Version]
- Lou, J.; Cao, G.; Li, R.; Liu, J.; Dong, Z.; Xu, L. β-Caryophyllene Attenuates Focal Cerebral Ischemia-Reperfusion Injury by Nrf2/HO-1 Pathway in Rats. Neurochem. Res. 2016, 41, 1291–1304. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, X.; Zhang, C.; Bai, X.; Zhang, J.; Zhao, X.; Chen, L.; Wang, L.; Zhu, C.; Cui, L.; et al. Nobiletin promotes antioxidant and anti-inflammatory responses and elicits protection against ischemic stroke in vivo. Brain Res. 2016, 1636, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Cai, J.; Kostuk, E.W.; Rosenwasser, R.; Iacovitti, L. Fumarate modulates the immune/inflammatory response and rescues nerve cells and neurological function after stroke in rats. J. Neuroinflamm. 2016, 13, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xian, J.W.; Choi, A.Y.-T.; Lau, C.B.-S.; Leung, W.N.; Ng, C.F.; Chan, C.W. Gastrodia and Uncaria (tianma gouteng) water extract exerts antioxidative and antiapoptotic effects against cerebral ischemia in vitro and in vivo. Chin. Med. 2016, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Chen, Y.; Yu, S.; Li, L.; Zhao, X.; Li, Q.; Zhao, J.; Zhao, Y. Neuroprotective effects of sulfiredoxin-1 during cerebral ischemia/reperfusion oxidative stress injury in rats. Brain Res. Bull. 2017, 132, 99–108. [Google Scholar] [CrossRef]
- Mršić-Pelčić, J.; Pilipović, K.; Pelčić, G.; Vitezić, D.; Župan, G. Decrease in Oxidative Stress Parameters after Post-Ischaemic Recombinant Human Erythropoietin Administration in the Hippocampus of Rats Exposed to Focal Cerebral Ischaemia. Basic Clin. Pharmacol. Toxicol. 2017, 121, 453–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atef, R.M.; Agha, A.M.; Abdel-Rhaman, A.-R.A.; Nassar, N.N. The Ying and Yang of Adenosine A1 and A2A Receptors on ERK1/2 Activation in a Rat Model of Global Cerebral Ischemia Reperfusion Injury. Mol. Neurobiol. 2018, 55, 1284–1298. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, Y.; He, Q.; Li, L.; Xie, H.; Zhao, Y.; Zhao, J. Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav. Brain Res. 2018, 336, 32–39. [Google Scholar] [CrossRef]
- Miao, Z.-Y.; Xia, X.; Che, L.; Song, Y.-T. Genistein attenuates brain damage induced by transient cerebral ischemia through up-regulation of Nrf2 expression in ovariectomized rats. Neurol. Res. 2018, 40, 689–695. [Google Scholar] [CrossRef]
- Guo, H.; Adah, D.; James, P.B.; Liu, Q.; Li, G.; Ahmadu, P.; Chai, L.; Wang, S.; Liu, Y.; Hu, L. Xueshuantong Injection (Lyophilized) Attenuates Cerebral Ischemia/Reperfusion Injury by the Activation of Nrf2–VEGF Pathway. Neurochem. Res. 2018, 43, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhao, Y.; Li, Y.; Wu, J.; Yu, S.; Zhu, J.; Li, L.; Zhao, Y. Sestrin2 overexpression attenuates focal cerebral ischemic injury in rat by increasing Nrf2/HO-1 pathway-mediated angiogenesis. Neuroscience 2019, 410, 140–149. [Google Scholar] [CrossRef]
- Li, Y.; Wu, J.; Yu, S.; Zhu, J.; Zhou, Y.; Wang, P.; Li, L.; Zhao, Y. Sestrin2 promotes angiogenesis to alleviate brain injury by activating Nrf2 through regulating the interaction between p62 and Keap1 following photothrombotic stroke in rats. Brain Res. 2020, 1745, 146948. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.; Shah, F.A.; Ali, T.; Tan, Z.; Alattar, A.; Ullah, N.; Khan, A.; Alshaman, R.; Li, S. Potent Natural Antioxidant Carveol Attenuates MCAO-Stress Induced Oxidative, Neurodegeneration by Regulating the Nrf-2 Pathway. Front. Neurosci. 2020, 14, 659. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, H.; Zhang, L.; Xu, Z.; Song, Y.; Wang, X.; Chu, R.; Xiao, Y.; Sun, M.; Ma, Y.; et al. Cottonseed Oil Alleviates Ischemic Stroke-Induced Oxidative Stress Injury Via Activating the Nrf2 Signaling Pathway. Mol. Neurobiol. 2021, 58, 2494–2507. [Google Scholar] [CrossRef]
- Gao, J.; Chen, N.; Li, N.; Xu, F.; Wang, W.; Lei, Y.; Shi, J.; Gong, Q. Neuroprotective Effects of Trilobatin, a Novel Naturally Occurring Sirt3 Agonist from Lithocarpus polystachyus Rehd., Mitigate Cerebral Ischemia/Reperfusion Injury: Involvement of TLR4/NF-κB and Nrf2/Keap-1 Signaling. Antioxid. Redox Signal. 2020, 33, 117–143. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yue, Y.; Li, J.; Li, Z.; Li, X.; Niu, Y.; Xiang, J.; Ding, H. Procyanidin B2 attenuates neurological deficits and blood-brain barrier disruption in a rat model of cerebral ischemia. Mol. Nutr. Food Res. 2015, 59, 1930–1941. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yue, Y.; Peng, A.; Zhang, L.; Xiang, J.; Cao, X.; Ding, H.; Yin, S. Myricetin ameliorates brain injury and neurological deficits via Nrf2 activation after experimental stroke in middle-aged rats. Food Funct. 2016, 7, 2624–2634. [Google Scholar] [CrossRef]
- Janyou, A.; Wicha, P.; Jittiwat, J.; Suksamrarn, A.; Tocharus, C.; Tocharus, J. Dihydrocapsaicin Attenuates Blood Brain Barrier and Cerebral Damage in Focal Cerebral Ischemia/Reperfusion via Oxidative Stress and Inflammatory. Sci. Rep. 2017, 7, 10556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.; Cui, J.; Yang, Z.; Guo, C.; Cao, J.; Xi, M.; Weng, Y.; Yin, Y.; Wang, Y.; Wei, G.; et al. Neuroprotective effect of Apelin 13 on ischemic stroke by activating AMPK/GSK-3β/Nrf2 signaling. J. Neuroinflamm. 2019, 16, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Li, X.; Wu, H.; Yang, Z.; Fei, L.; Zhu, J. Theaflavin attenuates cerebral ischemia/reperfusion injury by abolishing miRNA-128-3p-mediated Nrf2 inhibition and reducing oxidative stress. Mol. Med. Rep. 2019, 20, 4893–4904. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tucker, L.D.; Yan, D.; Lu, Y.; Yang, L.; Wu, C.; Li, Y.; Zhang, Q. Tert-butylhydroquinone post-treatment attenuates neonatal hypoxic-ischemic brain damage in rats. Neurochem. Int. 2018, 116, 1–12. [Google Scholar] [CrossRef]
- Liu, D.; Wang, H.; Zhang, Y.; Zhang, Z. Protective Effects of Chlorogenic Acid on Cerebral Ischemia/Reperfusion Injury Rats by Regulating Oxidative Stress-Related Nrf2 Pathway. Drug Des. Dev. Ther. 2020, 14, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kam, K.-Y.; Yu, S.J.; Jeong, N.; Hong, J.H.; Jalin, A.M.A.A.; Lee, S.; Choi, Y.W.; Lee, C.K.; Kang, S.G. p-Hydroxybenzyl alcohol prevents brain injury and behavioral impairment by activating Nrf2, PDI, and neurotrophic factor genes in a rat model of brain ischemia. Mol. Cells 2011, 31, 209–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Li, R.; Tu, P.; Chen, J.; Zeng, K.; Jiang, Y. Total Glycosides of Cistanche deserticola Promote Neurological Function Recovery by Inducing Neurovascular Regeneration via Nrf-2/Keap-1 Pathway in MCAO/R Rats. Front. Pharmacol. 2020, 11, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, T.-C. Nuclear Factor-Erythroid 2-Related Factor 2 (Nrf2) and Mitochondrial Dynamics/Mitophagy in Neurological Diseases. Antioxidants 2020, 9, 617. [Google Scholar] [CrossRef]
- Jiang, S.; Deng, C.; Lv, J.; Fan, C.; Hu, W.; Di, S.; Yan, X.; Ma, Z.; Liang, Z.; Yang, Y. Nrf2 Weaves an Elaborate Network of Neuroprotection Against Stroke. Mol. Neurobiol. 2017, 54, 1440–1455. [Google Scholar] [CrossRef]
- Li, H.; Tang, Z.; Chu, P.; Song, Y.; Yang, Y.; Sun, B.; Niu, M.; Qaed, E.; Shopit, A.; Han, G.; et al. Neuroprotective effect of phosphocreatine on oxidative stress and mitochondrial dysfunction induced apoptosis in vitro and in vivo: Involvement of dual PI3K/Akt and Nrf2/HO-1 pathways. Free Radic. Biol. Med. 2018, 120, 228–238. [Google Scholar] [CrossRef]
- Zolnourian, A.; Galea, I.; Bulters, D. Neuroprotective Role of the Nrf2 Pathway in Subarachnoid Haemorrhage and Its Therapeutic Potential. Oxid. Med. Cell. Longev. 2019, 2019, 1–21. [Google Scholar] [CrossRef]
- Narayanan, S.V.; Dave, K.R.; Saul, I.; Perez-Pinzon, M.A. Resveratrol Preconditioning Protects Against Cerebral Ischemic Injury via Nuclear Erythroid 2–Related Factor 2. Stroke 2015, 46, 1626–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, N.B.; Jain, S.; Agarwal, N.K.; Mediratta, P.K.; Sharma, K.K. Modulation of pentylenetetrazole-induced kindling and oxidative stress by curcumin in mice. Phytomedicine 2011, 18, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin Inhibits Formation of Amyloid β Oligomers and Fibrils, Binds Plaques, and Reduces Amyloid in Vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Bu, Q.; Liu, X.; Hu, W.; Wang, Y. Neuroprotective effect of TAT-14-3-3ε fusion protein against cerebral ischemia/reperfusion injury in rats. PLoS ONE 2014, 9, e93334. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.; Sanderson, I.R.; MacDonald, T.T. Curcumin as a therapeutic agent: The evidence from in vitro, animal and human studies. Br. J. Nutr. 2010, 103, 1545–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turpaev, K.T. Keap1-Nrf2 signaling pathway: Mechanisms of regulation and role in protection of cells against toxicity caused by xenobiotics and electrophiles. Biochem. Mosc. 2013, 78, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Chun, K.-S.; Kim, D.-H.; Kim, S.-J.; Kim, S.H.; Cho, N.-C.; Na, H.-K.; Surh, Y.-J. Curcumin induces stabilization of Nrf2 protein through Keap1 cysteine modification. Biochem. Pharmacol. 2020, 173, 113820. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Umar, S.; Ashafaq, M.; Akhtar, M.; Iqbal, Z.; Samim, M.; Ahmad, F.J. A comparative study of PNIPAM nanoparticles of curcumin, demethoxycurcumin, and bisdemethoxycurcumin and their effects on oxidative stress markers in experimental stroke. Protoplasma 2013, 250, 1327–1338. [Google Scholar] [CrossRef]
- Dohare, P.; Garg, P.; Jain, V.; Nath, C.; Ray, M. Dose dependence and therapeutic window for the neuroprotective effects of curcumin in thromboembolic model of rat. Behav. Brain Res. 2008, 193, 289–297. [Google Scholar] [CrossRef]
- Funk, J.L.; Frye, J.B.; Davis-Gorman, G.; Spera, A.L.; Bernas, M.J.; Witte, M.H.; Weinand, M.E.; Timmermann, B.N.; McDonagh, P.F.; Ritter, L. Curcuminoids Limit Neutrophil-Mediated Reperfusion Injury in Experimental Stroke by Targeting the Endothelium. Microcirculation 2013, 20, 544–554. [Google Scholar] [CrossRef]
- Ghoneim, A.I.; Abdel-Naim, A.B.; Khalifa, A.E.; El-Denshary, E.S. Protective effects of curcumin against ischaemia/reperfusion insult in rat forebrain. Pharmacol. Res. 2002, 46, 273–279. [Google Scholar] [CrossRef]
- Lapchak, P.A.; Schubert, D.R.; Maher, P.A. Delayed treatment with a novel neurotrophic compound reduces behavioral deficits in rabbit ischemic stroke: Neuroprotection from ischemic stroke. J. Neurochem. 2011, 116, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Rathore, P.; Dohare, P.; Varma, S.; Ray, A.; Sharma, U.; Jaganathanan, N.R.; Ray, M. Curcuma Oil: Reduces Early Accumulation of Oxidative Product and is Anti-apoptogenic in Transient Focal Ischemia in Rat Brain. Neurochem. Res. 2008, 33, 1672–1682. [Google Scholar] [CrossRef]
- Shukla, P.K.; Khanna, V.K.; Ali, M.M.; Khan, M.Y.; Srimal, R.C. Anti-ischemic effect of curcumin in rat brain. Neurochem. Res. 2008, 33, 1036–1043. [Google Scholar] [CrossRef]
- Thiyagarajan, M.; Sharma, S.S. Neuroprotective effect of curcumin in middle cerebral artery occlusion induced focal cerebral ischemia in rats. Life Sci. 2004, 74, 969–985. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yu, S.; Zheng, W.; Feng, G.; Luo, G.; Wang, L.; Zhao, Y. Curcumin improves outcomes and attenuates focal cerebral ischemic injury via antiapoptotic mechanisms in rats. Neurochem. Res. 2010, 35, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, W.; Sun, Y.J.; Hu, M.; Li, F.; Zhu, D.Y. Neuroprotective effect of curcumin on focal cerebral ischemic rats by preventing blood-brain barrier damage. Eur. J. Pharmacol. 2007, 561, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Suwanwela, N.C.; Patumraj, S. Curcumin by down-regulating NF-kB and elevating Nrf2, reduces brain edema and neurological dysfunction after cerebral I/R. Microvasc. Res. 2016, 106, 117–127. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, Y.; Zheng, W.; Lu, Y.; Feng, G.; Yu, S. Neuroprotective effect of curcumin on transient focal cerebral ischemia in rats. Brain Res. 2008, 1229, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, M.A.; Waeber, C. Remote ischemic preconditioning: Making the brain more tolerant, safely and inexpensively. Circulation 2011, 123, 709–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Reyes, S.; Guzmán-Beltrán, S.; Medina-Campos, O.N.; Pedraza-Chaverri, J. Curcumin Pretreatment Induces Nrf2 and an Antioxidant Response and Prevents Hemin-Induced Toxicity in Primary Cultures of Cerebellar Granule Neurons of Rats. Oxid. Med. Cell. Longev. 2013, 2013, 1–14. [Google Scholar] [CrossRef]
- Zhao, R.; Yang, B.; Wang, L.; Xue, P.; Deng, B.; Zhang, G.; Jiang, S.; Zhang, M.; Liu, M.; Pi, J.; et al. Curcumin protects human keratinocytes against inorganic arsenite-induced acute cytotoxicity through an NRF2-dependent mechanism. Oxid. Med. Cell. Longev. 2013, 2013, 412576. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, X.; Fan, H.; Liu, Y. Curcumin upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. 2009, 1282, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.N.; Robinson, S.R.; Dringen, R.; Bishop, G.M. Uptake, metabolism and toxicity of hemin in cultured neurons. Neurochem. Int. 2011, 58, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Liu, Z.; Liang, G. Promising curcumin-based drug design: Mono-carbonyl analogues of curcumin (MACs). Curr. Pharm. Des. 2013, 19, 2114–2135. [Google Scholar] [PubMed]
- Jangra, A.; Kwatra, M.; Singh, T.; Pant, R.; Kushwah, P.; Sharma, Y.; Saroha, B.; Datusalia, A.K.; Bezbaruah, B.K. Piperine Augments the Protective Effect of Curcumin Against Lipopolysaccharide-Induced Neurobehavioral and Neurochemical Deficits in Mice. Inflammation 2016, 39, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Antony, B.; Merina, B.; Iyer, V.; Judy, N.; Lennertz, K.; Joyal, S. A pilot cross-over study to evaluate human oral bioavailability of BCM-95® CG (BiocurcumaxTM), a novel bioenhanced preparation of curcumin. Indian J. Pharm. Sci. 2008, 70, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werdenberg, D.; Joshi, R.; Wolffram, S.; Merkle, H.P.; Langguth, P. Presystemic metabolism and intestinal absorption of antipsoriatic fumaric acid esters. Biopharm. Drug Dispos. 2003, 24, 259–273. [Google Scholar] [CrossRef]
- Linker, R.A.; Haghikia, A. Dimethyl fumarate in multiple sclerosis: Latest developments, evidence and place in therapy. Ther. Adv. Chronic Dis. 2016, 7, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Reich, K.; Thaci, D.; Mrowietz, U.; Kamps, A.; Neureither, M.; Luger, T. Efficacy and safety of fumaric acid esters in the long-term treatment of psoriasis—A retrospective study (FUTURE). J. Dtsch. Dermatol. Ges. 2009, 7, 603–611. [Google Scholar] [CrossRef]
- Meissner, M.; Valesky, E.M.; Kippenberger, S.; Kaufmann, R. Dimethyl fumarate—Only an anti-psoriatic medication? J. Dtsch. Dermatol. Ges. 2012, 10, 793–801. [Google Scholar] [CrossRef]
- Fox, R.J.; Miller, D.H.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Yang, M.; Raghupathi, K.; Novas, M.; Sweetser, M.T.; et al. Placebo-Controlled Phase 3 Study of Oral BG-12 or Glatiramer in Multiple Sclerosis. N. Engl. J. Med. 2012, 367, 1087–1097. [Google Scholar] [CrossRef] [Green Version]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-Controlled Phase 3 Study of Oral BG-12 for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef] [Green Version]
- Salmen, A.; Gold, R. Mode of action and clinical studies with fumarates in multiple sclerosis. Exp. Neurol. 2014, 262 Pt A, 52–56. [Google Scholar] [CrossRef]
- De Jong, R.; Bezemer, A.C.; Zomerdijk, T.P.L.; van de Pouw-Kraan, T.; Ottenhoff, T.H.M.; Nibbering, P.H. Selective stimulation of T helper 2 cytokine responses by the anti-psoriasis agent monomethylfumarate. Eur. J. Immunol. 1996, 26, 2067–2074. [Google Scholar] [CrossRef]
- Asadullah, K.; Schmid, H.; Friedrich, M.; Randow, F.; Volk, H.-D.; Sterry, W.; Döcke, W.-D. Influence of monomethylfumarate on monocytic cytokine formation—Explanation for adverse and therapeutic effects in psoriasis? Arch. Dermatol. Res. 1997, 289, 623–630. [Google Scholar] [CrossRef]
- Litjens, N.H.R.; Rademaker, M.; Ravensbergen, B.; Rea, D.; van der Plas, M.J.A.; Thio, B.; Walding, A.; van Dissel, J.T.; Nibbering, P.H. Monomethylfumarate affects polarization of monocyte-derived dendritic cells resulting in down-regulated Th1 lymphocyte responses. Eur. J. Immunol. 2004, 34, 565–575. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Brück, J.; Kellerer, C.; Deng, C.; Peng, H.; Rothfuss, O.; Hussain, R.Z.; Gocke, A.R.; Respa, A.; Glocova, I.; et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J. Exp. Med. 2011, 208, 2291–2303. [Google Scholar] [CrossRef]
- Treumer, F.; Zhu, K.; Gläser, R.; Mrowietz, U. Dimethylfumarate is a potent inducer of apoptosis in human T cells. J. Investig. Dermatol. 2003, 121, 1383–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loewe, R.; Holnthoner, W.; Gröger, M.; Pillinger, M.; Gruber, F.; Mechtcheriakova, D.; Hofer, E.; Wolff, K.; Petzelbauer, P. Dimethylfumarate inhibits TNF-induced nuclear entry of NF-kappa B/p65 in human endothelial cells. J. Immunol. 2002, 168, 4781–4787. [Google Scholar] [CrossRef] [PubMed]
- Nibbering, P.H.; Thio, B.; Zomerdijk, T.P.; Bezemer, A.C.; Beijersbergen, R.L.; van Furth, R. Effects of monomethylfumarate on human granulocytes. J. Investig. Dermatol. 1993, 101, 37–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ockenfels, H.M.; Schultewolter, T.; Ockenfels, G.; Funk, R.; Goos, M. The antipsoriatic agent dimethylfumarate immunomodulates T-cell cytokine secretion and inhibits cytokines of the psoriatic cytokine network. Br. J. Dermatol. 1998, 139, 390–395. [Google Scholar] [CrossRef]
- Schilling, S.; Goelz, S.; Linker, R.; Luehder, F.; Gold, R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin. Exp. Immunol. 2006, 145, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Sebok, B.; Bonnekoh, B.; Vetter, R.; Schneider, I.; Gollnick, H.; Mahrle, G. The antipsoriatic dimethyl-fumarate suppresses interferon-gamma -induced ICAM-1 and HLA-DR expression on hyperproliferative keratinocytes. Quantification by a culture plate-directed APAAP-ELISA technique. Eur. J. Dermatol. 1998, 8, 29–32. [Google Scholar] [PubMed]
- Stoof, T.J.; Flier, J.; Sampat, S.; Nieboer, C.; Tensen, C.P.; Boorsma, D.M. The antipsoriatic drug dimethylfumarate strongly suppresses chemokine production in human keratinocytes and peripheral blood mononuclear cells. Br. J. Dermatol. 2001, 144, 1114–1120. [Google Scholar] [CrossRef]
- Linker, R.A.; Lee, D.-H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [Green Version]
- Scannevin, R.H.; Chollate, S.; Jung, M.; Shackett, M.; Patel, H.; Bista, P.; Zeng, W.; Ryan, S.; Yamamoto, M.; Lukashev, M.; et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther. 2012, 341, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.X.; Lisi, L.; Dello Russo, C.; Polak, P.E.; Sharp, A.; Weinberg, G.; Kalinin, S.; Feinstein, D.L. The anti-inflammatory effects of dimethyl fumarate in astrocytes involve glutathione and haem oxygenase-1. ASN Neuro 2011, 3, e00055. [Google Scholar] [CrossRef] [PubMed]
- Iniaghe, L.O.; Krafft, P.R.; Klebe, D.W.; Omogbai, E.K.I.; Zhang, J.H.; Tang, J. Dimethyl fumarate confers neuroprotection by casein kinase 2 phosphorylation of Nrf2 in murine intracerebral hemorrhage. Neurobiol. Dis. 2015, 82, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Sun, G.; Zhang, J.; Ting, S.-M.; Gonzales, N.; Aronowski, J. Dimethyl Fumarate Protects Brain From Damage Produced by Intracerebral Hemorrhage by Mechanism Involving Nrf2. Stroke 2015, 46, 1923–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunze, R.; Urrutia, A.; Hoffmann, A.; Liu, H.; Helluy, X.; Pham, M.; Reischl, S.; Korff, T.; Marti, H.H. Dimethyl fumarate attenuates cerebral edema formation by protecting the blood-brain barrier integrity. Exp. Neurol. 2015, 266, 99–111. [Google Scholar] [CrossRef]
- Lin-Holderer, J.; Li, L.; Gruneberg, D.; Marti, H.H.; Kunze, R. Fumaric acid esters promote neuronal survival upon ischemic stress through activation of the Nrf2 but not HIF-1 signaling pathway. Neuropharmacology 2016, 105, 228–240. [Google Scholar] [CrossRef]
- Liu, L.; Vollmer, M.K.; Ahmad, A.S.; Fernandez, V.M.; Kim, H.; Doré, S. Pretreatment with Korean red ginseng or dimethyl fumarate attenuates reactive gliosis and confers sustained neuroprotection against cerebral hypoxic-ischemic damage by an Nrf2-dependent mechanism. Free Radic. Biol. Med. 2019, 131, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Vollmer, M.K.; Kelly, M.G.; Fernandez, V.M.; Fernandez, T.G.; Kim, H.; Doré, S. Reactive Gliosis Contributes to Nrf2-Dependent Neuroprotection by Pretreatment with Dimethyl Fumarate or Korean Red Ginseng Against Hypoxic-Ischemia: Focus on Hippocampal Injury. Mol. Neurobiol. 2020, 57, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, T.; Liu, H.; Wang, X.; Bo, S.; Xie, Y.; Bai, X.; Wu, L.; Wang, Z.; Liu, D. Resveratrol exerts antidepressant properties in the chronic unpredictable mild stress model through the regulation of oxidative stress and mTOR pathway in the rat hippocampus and prefrontal cortex. Behav. Brain Res. 2016, 302, 191–199. [Google Scholar] [CrossRef]
- Wang, L.; Wang, C.; Jia, Y.; Liu, Z.; Shu, X.; Liu, K. Resveratrol Increases Anti-Proliferative Activity of Bestatin Through Downregulating P-Glycoprotein Expression Via Inhibiting PI3K/Akt/mTOR Pathway in K562/ADR Cells. J. Cell. Biochem. 2016, 117, 1233–1239. [Google Scholar] [CrossRef]
- Sui, T.; Ma, L.; Bai, X.; Li, Q.; Xu, X. Resveratrol inhibits the phosphatidylinositide 3-kinase/protein kinase B/mammalian target of rapamycin signaling pathway in the human chronic myeloid leukemia K562 cell line. Oncol. Lett. 2014, 7, 2093–2098. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.-H.; Lee, H.; Lee, S.-R. Protective effect of resveratrol against neuronal damage following transient global cerebral ischemia in mice. J. Nutr. Biochem. 2016, 27, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Li, Y.-H.; Wang, J.-J.; Pan, J.; Lu, H. Endoplasmic reticulum stress could induce autophagy and apoptosis and enhance chemotherapy sensitivity in human esophageal cancer EC9706 cells by mediating PI3K/Akt/mTOR signaling pathway. Tumour Biol. 2017, 39, 101042831770574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simão, F.; Matté, A.; Pagnussat, A.S.; Netto, C.A.; Salbego, C.G. Resveratrol prevents CA1 neurons against ischemic injury by parallel modulation of both GSK-3β and CREB through PI3-K/Akt pathways: Resveratrol prevents ischemic injury through PI3-K. Eur. J. Neurosci. 2012, 36, 2899–2905. [Google Scholar] [CrossRef]
- Singh, N.; Agrawal, M.; Doré, S. Neuroprotective properties and mechanisms of resveratrol in in vitro and in vivo experimental cerebral stroke models. ACS Chem. Neurosci. 2013, 4, 1151–1162. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, M.; Kumar, V.; Singh, A.K.; Kashyap, M.P.; Khanna, V.K.; Siddiqui, M.A.; Pant, A.B. trans -Resveratrol Protects Ischemic PC12 Cells by Inhibiting the Hypoxia Associated Transcription Factors and Increasing the Levels of Antioxidant Defense Enzymes. ACS Chem. Neurosci. 2013, 4, 285–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamin, L.L.; Dillenburg-Pilla, P.; Argenta-Comiran, R.; Horn, A.P.; Simão, F.; Nassif, M.; Gerhardt, D.; Frozza, R.L.; Salbego, C. Protective effect of resveratrol against oxygen–glucose deprivation in organotypic hippocampal slice cultures: Involvement of PI3-K pathway. Neurobiol. Dis. 2006, 24, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Bournival, J.; Quessy, P.; Martinoli, M.-G. Protective Effects of Resveratrol and Quercetin Against MPP+ -Induced Oxidative Stress Act by Modulating Markers of Apoptotic Death in Dopaminergic Neurons. Cell Mol. Neurobiol. 2009, 29, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Zini, R.; Morin, C.; Bertelli, A.; Bertelli, A.A.; Tillement, J.P. Effects of resveratrol on the rat brain respiratory chain. Drugs Exp. Clin. Res. 1999, 25, 87–97. [Google Scholar]
- Hou, Y.; Wang, K.; Wan, W.; Cheng, Y.; Pu, X.; Ye, X. Resveratrol provides neuroprotection by regulating the JAK2/STAT3/PI3K/AKT/mTOR pathway after stroke in rats. Genes Dis. 2018, 5, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E.; de Oliveira, A.; Gräf, S.; Bhatia, H.S.; Hüll, M.; Muñoz, E.; Fiebich, B.L. Resveratrol potently reduces prostaglandin E2 production and free radical formation in lipopolysaccharide-activated primary rat microglia. J. Neuroinflamm. 2007, 4, 25. [Google Scholar] [CrossRef] [Green Version]
- Bi, X.L.; Yang, J.Y.; Dong, Y.X.; Wang, J.M.; Cui, Y.H.; Ikeshima, T.; Zhao, Y.Q.; Wu, C.F. Resveratrol inhibits nitric oxide and TNF-α production by lipopolysaccharide-activated microglia. Int. Immunopharmacol. 2005, 5, 185–193. [Google Scholar] [CrossRef]
- Kim, Y.A.; Kim, G.-Y.; Park, K.-Y.; Choi, Y.H. Resveratrol Inhibits Nitric Oxide and Prostaglandin E2 Production by Lipopolysaccharide-Activated C6 Microglia. J. Med. Food 2007, 10, 218–224. [Google Scholar] [CrossRef]
- Bureau, G.; Longpré, F.; Martinoli, M.-G. Resveratrol and quercetin, two natural polyphenols, reduce apoptotic neuronal cell death induced by neuroinflammation. J. Neurosci. Res. 2008, 86, 403–410. [Google Scholar] [CrossRef]
- Innamorato, N.G.; Rojo, A.I.; García-Yagüe, Á.J.; Yamamoto, M.; de Ceballos, M.L.; Cuadrado, A. The Transcription Factor Nrf2 Is a Therapeutic Target against Brain Inflammation. J. Immunol. 2008, 181, 680–689. [Google Scholar] [CrossRef] [Green Version]
- Shah, Z.A.; Li, R.-C.; Thimmulappa, R.K.; Kensler, T.W.; Yamamoto, M.; Biswal, S.; Doré, S. Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience 2007, 147, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Jang, J.-H.; Li, M.-H.; Surh, Y.-J. Resveratrol upregulates heme oxygenase-1 expression via activation of NF-E2-related factor 2 in PC12 cells. Biochem. Biophys. Res. Commun. 2005, 331, 993–1000. [Google Scholar] [CrossRef]
- Agrawal, M.; Kumar, V.; Kashyap, M.P.; Khanna, V.K.; Randhawa, G.S.; Pant, A.B. Ischemic insult induced apoptotic changes in PC12 cells: Protection by trans resveratrol. Eur. J. Pharmacol. 2011, 666, 5–11. [Google Scholar] [CrossRef]
- Ungvari, Z.; Bagi, Z.; Feher, A.; Recchia, F.A.; Sonntag, W.E.; Pearson, K.; de Cabo, R.; Csiszar, A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H18–H24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Huang, J.; Shen, C.; Cheng, W.; Yu, P.; Wang, L.; Tang, F.; Guo, S.; Yang, Q.; Zhang, J. Resveratrol Treatment in Different Time-Attenuated Neuronal Apoptosis After Oxygen and Glucose Deprivation/Reoxygenation via Enhancing the Activation of Nrf-2 Signaling Pathway In Vitro. Cell Transplant. 2018, 27, 1789–1797. [Google Scholar] [CrossRef]
- Van Horssen, J.; Schreibelt, G.; Bö, L.; Montagne, L.; Drukarch, B.; van Muiswinkel, F.L.; de Vries, H.E. NAD(P)H:quinone oxidoreductase 1 expression in multiple sclerosis lesions. Free Radic. Biol. Med. 2006, 41, 311–317. [Google Scholar] [CrossRef]
- Ghawi, S.K.; Methven, L.; Niranjan, K. The potential to intensify sulforaphane formation in cooked broccoli (Brassica oleracea var. italica) using mustard seeds (Sinapis alba). Food Chem. 2013, 138, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.-S.; Jeffery, E.H. Induction of Quinone Reductase by Sulforaphane and Sulforaphane N -Acetylcysteine Conjugate in Murine Hepatoma Cells. J. Med. Food 2005, 8, 198–203. [Google Scholar] [CrossRef]
- Clarke, J.D.; Dashwood, R.H.; Ho, E. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 2008, 269, 291–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Chen, G.-D.; Zhou, X.; McGinnis, J.F.; Li, F.; Cao, W. Molecular mechanisms underlying cochlear degeneration in the tubby mouse and the therapeutic effect of sulforaphane. Neurochem. Int. 2009, 54, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Talalay, P.; Fahey, J.W.; Healy, Z.R.; Wehage, S.L.; Benedict, A.L.; Min, C.; Dinkova-Kostova, A.T. Sulforaphane mobilizes cellular defenses that protect skin against damage by UV radiation. Proc. Natl. Acad. Sci. USA 2007, 104, 17500–17505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shokeir, A.A.; Barakat, N.; Hussein, A.M.; Awadalla, A.; Harraz, A.M.; Khater, S.; Hemmaid, K.; Kamal, A.I. Activation of Nrf2 by Ischemic Preconditioning and Sulforaphane in Renal Ischemia/Reperfusion Injury: A Comparative Experimental Study. Physiol Res. 2015, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; He, M.; Liu, R.; Brecha, N.C.; Yu, A.C.H.; Pu, M. Sulforaphane Protects Rodent Retinas against Ischemia-Reperfusion Injury through the Activation of the Nrf2/HO-1 Antioxidant Pathway. PLoS ONE 2014, 9, e114186. [Google Scholar] [CrossRef]
- Chen, Z.; Mohr, A.; Heitplatz, B.; Hansen, U.; Pascher, A.; Brockmann, J.G.; Becker, F. Sulforaphane Elicits Protective Effects in Intestinal Ischemia Reperfusion Injury. Int. J. Mol. Sci. 2020, 21, 5189. [Google Scholar] [CrossRef]
- Zhao, H.-D. Sulforaphane protects liver injury induced by intestinal ischemia reperfusion through Nrf2-ARE pathway. WJG 2010, 16, 3002. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Kobori, N.; Aronowski, J.; Dash, P.K. Sulforaphane reduces infarct volume following focal cerebral ischemia in rodents. Neurosci. Lett. 2006, 393, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Moore, A.N.; Clifton, G.L.; Dash, P.K. Sulforaphane enhances aquaporin-4 expression and decreases cerebral edema following traumatic brain injury. J. Neurosci. Res. 2005, 82, 499–506. [Google Scholar] [CrossRef]
- Ma, L.-L.; Xing, G.-P.; Yu, Y.; Liang, H.; Yu, T.-X.; Zheng, W.-H.; Lai, T.-B. Sulforaphane exerts neuroprotective effects via suppression of the inflammatory response in a rat model of focal cerebral ischemia. Int. J. Clin. Exp. Med. 2015, 8, 17811–17817. [Google Scholar]
- Tarozzi, A.; Angeloni, C.; Malaguti, M.; Morroni, F.; Hrelia, S.; Hrelia, P. Sulforaphane as a Potential Protective Phytochemical against Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2013, 2013, 1–10. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, T.; Mao, L.; Zhang, F. Sulforaphane Protects against Brain Diseases: Roles of Cytoprotective Enzymes. Austin J. Cerebrovasc. Dis. Stroke 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Ping, Z.; Liu, W.; Kang, Z.; Cai, J.; Wang, Q.; Cheng, N.; Wang, S.; Wang, S.; Zhang, J.H.; Sun, X. Sulforaphane protects brains against hypoxic–ischemic injury through induction of Nrf2-dependent phase 2 enzyme. Brain Res. 2010, 1343, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Danilov, C.A.; Chandrasekaran, K.; Racz, J.; Soane, L.; Zielke, C.; Fiskum, G. Sulforaphane protects astrocytes against oxidative stress and delayed death caused by oxygen and glucose deprivation. Glia 2009, 57, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Cui, W.; Xin, Y.; Miao, X.; Barati, M.T.; Zhang, C.; Chen, Q.; Tan, Y.; Cui, T.; Zheng, Y.; et al. Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of Nrf2 expression and transcription activation. J. Mol. Cell. Cardiol. 2013, 57, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.-Y.; Zhang, C.; Lee, J.H.; Shu, L.; Wu, T.-Y.; Khor, T.O.; Conney, A.H.; Lu, Y.-P.; Kong, A.-N.T. Requirement and Epigenetics Reprogramming of Nrf2 in Suppression of Tumor Promoter TPA-Induced Mouse Skin Cell Transformation by Sulforaphane. Cancer Prev. Res. 2014, 7, 319–329. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.; Lamont, D.J.; Beattie, K.A.; Hayes, J.D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. USA 2010, 107, 18838–18843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent Proteasomal Degradation of Transcription Factor Nrf2 Contributes to the Negative Regulation of Antioxidant Response Element-driven Gene Expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Yang, T.; Li, X.; Lei, X.; Sun, Y.; Zhao, Y.; Zhang, W.; Gao, Y.; Sun, B.; Zhang, F. Protective effects of sulforaphane in experimental vascular cognitive impairment: Contribution of the Nrf2 pathway. J. Cereb. Blood Flow Metab. 2019, 39, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Egner, P.A.; Chen, J.G.; Wang, J.B.; Wu, Y.; Sun, Y.; Lu, J.H.; Zhu, J.; Zhang, Y.H.; Chen, Y.S.; Friesen, M.D.; et al. Bioavailability of Sulforaphane from Two Broccoli Sprout Beverages: Results of a Short-term, Cross-over Clinical Trial in Qidong, China. Cancer Prev. Res. 2011, 4, 384–395. [Google Scholar] [CrossRef] [Green Version]
- Hu, R.; Hebbar, V.; Kim, B.-R.; Chen, C.; Winnik, B.; Buckley, B.; Soteropoulos, P.; Tolias, P.; Hart, R.P.; Kong, A.-N.T. In Vivo Pharmacokinetics and Regulation of Gene Expression Profiles by Isothiocyanate Sulforaphane in the Rat. J. Pharmacol. Exp. Ther. 2004, 310, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Dinkova-Kostova, A.T.; Wade, K.L.; Zhang, Y.; Shapiro, T.A.; Talalay, P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: Pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin. Chim. Acta 2002, 316, 43–53. [Google Scholar] [CrossRef]
- Jazwa, A.; Rojo, A.I.; Innamorato, N.G.; Hesse, M.; Fernández-Ruiz, J.; Cuadrado, A. Pharmacological Targeting of the Transcription Factor Nrf2 at the Basal Ganglia Provides Disease Modifying Therapy for Experimental Parkinsonism. Antioxid. Redox Signal. 2011, 14, 2347–2360. [Google Scholar] [CrossRef] [Green Version]
- Clarke, J.D.; Hsu, A.; Williams, D.E.; Dashwood, R.H.; Stevens, J.F.; Yamamoto, M.; Ho, E. Metabolism and Tissue Distribution of Sulforaphane in Nrf2 Knockout and Wild-Type Mice. Pharm. Res. 2011, 28, 3171–3179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Toxicology Program. NTP Toxicology and Carcinogenesis Studies of t-Butylhydroquinone (CAS No. 1948-33-0) in F344/N Rats and B6C3F(1) Mice (Feed Studies). Natl. Toxicol. Program. Tech. Rep. Ser. 1997, 459, 1–326. [Google Scholar]
- Joint FAO/WHO Expert Committee on Food Additives (Ed.) Evaluation of Certain Food Additives and Contaminants: Forty-Ninth Report of the Joint FAO/WHO Expert Committee on Food Additives; WHO Technical Report Series; World Health Organization: Geneva, Switzerland, 1999; ISBN 978-92-4-120884-0. [Google Scholar]
- Saykally, J.N.; Rachmany, L.; Hatic, H.; Shaer, A.; Rubovitch, V.; Pick, C.G.; Citron, B.A. The nuclear factor erythroid 2-like 2 activator, tert-butylhydroquinone, improves cognitive performance in mice after mild traumatic brain injury. Neuroscience 2012, 223, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ji, C.; Wu, L.; Qiu, J.; Li, Q.; Shao, Z.; Chen, G. Tert-Butylhydroquinone Alleviates Early Brain Injury and Cognitive Dysfunction after Experimental Subarachnoid Hemorrhage: Role of Keap1/Nrf2/ARE Pathway. PLoS ONE 2014, 9, e97685. [Google Scholar] [CrossRef] [Green Version]
- De Long, M.J.; Santamaria, A.B.; Talalay, P. Role of cytochrome P 1 in the induction of NAD(P)H:quinone reductase in a murine hepatoma cell line and its mutants. Carcinogenesis 1987, 8, 1549–1553. [Google Scholar] [CrossRef]
- Talalay, P. Mechanisms of induction of enzymes that protect against chemical carcinogenesis. Adv. Enzym. Regul. 1989, 28, 237–250. [Google Scholar] [CrossRef]
- Van Ommen, B.; Koster, A.; Verhagen, H.; van Bladeren, P.J. The glutathione conjugates of tert-butyl hydroquinone as potent redox cycling agents and possible reactive agents underlying the toxicity of butylated hydroxyanisole. Biochem. Biophys. Res. Commun. 1992, 189, 309–314. [Google Scholar] [CrossRef]
- Gharavi, N.; Haggarty, S.; El-Kadi, A.O.S. Chemoprotective and Carcinogenic Effects of tert-Butylhydroquinone and Its Metabolites. CDM 2007, 8, 1–7. [Google Scholar] [CrossRef]
- Peters, M.M.; Rivera, M.I.; Jones, T.W.; Monks, T.J.; Lau, S.S. Glutathione conjugates of tert-butyl-hydroquinone, a metabolite of the urinary tract tumor promoter 3-tert-butyl-hydroxyanisole, are toxic to kidney and bladder. Cancer Res. 1996, 56, 1006–1011. [Google Scholar]
- Sun, J.; Hu, H.; Ren, X.; Simpkins, J.W. Tert-butylhydroquinone compromises survival in murine experimental stroke. Neurotoxicol. Teratol. 2016, 54, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev. Pharmacol. Toxicol 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Zeynalov, E.; Doré, S. Low doses of carbon monoxide protect against experimental focal brain ischemia. Neurotox Res. 2009, 15, 133–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otterbein, L.E. Carbon monoxide: Innovative anti-inflammatory properties of an age-old gas molecule. Antioxid. Redox Signal. 2002, 4, 309–319. [Google Scholar] [CrossRef]
- Leffler, C.W.; Parfenova, H.; Jaggar, J.H.; Wang, R. Carbon monoxide and hydrogen sulfide: Gaseous messengers in cerebrovascular circulation. J. Appl. Physiol. 2006, 100, 1065–1076. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-H.; Tsai, H.-L.; Chiang, M.-T.; Chau, L.-Y. Carbon Monoxide-Induced Early Thrombolysis Contributes to Heme Oxygenase-1-Mediated Inhibition of Neointimal Growth after Vascular Injury in Hypercholesterolemic Mice. J. Biomed. Sci. 2006, 13, 721–730. [Google Scholar] [CrossRef]
- Liao, S.; Wu, J.; Liu, R.; Wang, S.; Luo, J.; Yang, Y.; Qin, Y.; Li, T.; Zheng, X.; Song, J.; et al. A novel compound DBZ ameliorates neuroinflammation in LPS-stimulated microglia and ischemic stroke rats: Role of Akt(Ser473)/GSK3β(Ser9)-mediated Nrf2 activation. Redox Biol. 2020, 36, 101644. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, W.; Matei, N.; Li, X.; Pang, J.; Mo, J.; Chen, S.; Tang, J.; Yan, M.; Zhang, J.H. Ezetimibe Attenuates Oxidative Stress and Neuroinflammation via the AMPK/Nrf2/TXNIP Pathway after MCAO in Rats. Oxid. Med. Cell. Longev. 2020, 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Lu, H.; Qin, J.; Qu, S.; Wang, W.; Guo, Y.; Liao, W.; Song, M.; Chen, J.; Wang, Y. Biochanin A Provides Neuroprotection Against Cerebral Ischemia/Reperfusion Injury by Nrf2-Mediated Inhibition of Oxidative Stress and Inflammation Signaling Pathway in Rats. Med. Sci. Monit. 2019, 25, 8975–8983. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhang, H.; Zhang, J.; Yan, M. Isoquercetin attenuates oxidative stress and neuronal apoptosis after ischemia/reperfusion injury via Nrf2-mediated inhibition of the NOX4/ROS/NF-κB pathway. Chem. Biol. Interact. 2018, 284, 32–40. [Google Scholar] [CrossRef]
- Ma, T.; Shi, Y.; Wang, Y. Forsythiaside A protects against focal cerebral ischemic injury by mediating the activation of the Nrf2 and endoplasmic reticulum stress pathways. Mol. Med. Rep. 2019, 20, 1313–1320. [Google Scholar] [CrossRef]
- Chen, L.; Wang, L.; Zhang, X.; Cui, L.; Xing, Y.; Dong, L.; Liu, Z.; Li, Y.; Zhang, X.; Wang, C.; et al. The protection by Octreotide against experimental ischemic stroke: Up-regulated transcription factor Nrf2, HO-1 and down-regulated NF-κB expression. Brain Res. 2012, 1475, 80–87. [Google Scholar] [CrossRef]
- Zhou, F.; Wang, M.; Ju, J.; Wang, Y.; Liu, Z.; Zhao, X.; Yan, Y.; Yan, S.; Luo, X.; Fang, Y. Schizandrin A protects against cerebral ischemia-reperfusion injury by suppressing inflammation and oxidative stress and regulating the AMPK/Nrf2 pathway regulation. Am. J. Transl. Res. 2019, 11, 199–209. [Google Scholar] [PubMed]
- Xie, Y.; Zhang, X.; Zhang, C.; Yang, Y.; He, J.; Chen, Y. Protective effects of leonurine against ischemic stroke in mice by activating nuclear factor erythroid 2-related factor 2 pathway. CNS Neurosci. Ther. 2019, 25, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Song, J.; Yan, R.; Li, L.; Xiao, Z.; Zhou, W.; Wang, Z.; Xiao, W.; Du, G. Diterpene ginkgolides protect against cerebral ischemia/reperfusion damage in rats by activating Nrf2 and CREB through PI3K/Akt signaling. Acta Pharmacol. Sin. 2018, 39, 1259–1272. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, X.; Chang, S.; Wang, Y.; Xu, Y.; Ran, S.; Huang, Z.; Li, P.; Li, J.; Zhang, L.; et al. Totarol prevents neuronal injury in vitro and ameliorates brain ischemic stroke: Potential roles of Akt activation and HO-1 induction. Toxicol. Appl. Pharmacol. 2015, 289, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Maharjan, S.; Wang, Q.; Sun, Y.; Han, X.; Wang, S.; Mao, Z.; Xin, Y.; Zhang, B. α-Lipoic Acid Promotes Neurological Recovery After Ischemic Stroke by Activating the Nrf2/HO-1 Pathway to Attenuate Oxidative Damage. Cell. Physiol. Biochem. 2017, 43, 1273–1287. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, S.; Mao, L.; Leak, R.K.; Shi, Y.; Zhang, W.; Hu, X.; Sun, B.; Cao, G.; Gao, Y.; et al. Omega-3 Fatty Acids Protect the Brain against Ischemic Injury by Activating Nrf2 and Upregulating Heme Oxygenase 1. J. Neurosci. 2014, 34, 1903–1915. [Google Scholar] [CrossRef]
- Wei, C.-C.; Kong, Y.-Y.; Li, G.-Q.; Guan, Y.-F.; Wang, P.; Miao, C.-Y. Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO-1 signaling pathway. Sci. Rep. 2017, 7, 717. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Zhou, D.; Yan, B. Eriocitrin alleviates oxidative stress and inflammatory response in cerebral ischemia reperfusion rats by regulating phosphorylation levels of Nrf2/NQO-1/HO-1/NF-κB p65 proteins. Ann. Transl. Med. 2020, 8, 757. [Google Scholar] [CrossRef] [PubMed]
- Yen, T.-L.; Chen, R.-J.; Jayakumar, T.; Lu, W.-J.; Hsieh, C.-Y.; Hsu, M.-J.; Yang, C.-H.; Chang, C.-C.; Lin, Y.-K.; Lin, K.-H.; et al. Andrographolide stimulates p38 mitogen-activated protein kinase–nuclear factor erythroid-2-related factor 2–heme oxygenase 1 signaling in primary cerebral endothelial cells for definite protection against ischemic stroke in rats. Transl. Res. 2016, 170, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Yang, Q.; Yang, B.; Xu, H.; Nasif, O.; Muruganantham, S.; Chen, J. Phyllanthin Averts Oxidative Stress and Neuroinflammation in Cerebral Ischemic-Reperfusion Injury through Modulation of the NF-κB and AMPK/Nrf2 Pathways. J. Environ. Pathol. Toxicol. Oncol. 2021, 40, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-J.; Cui, P. Neohesperidin attenuates cerebral ischemia–reperfusion injury via inhibiting the apoptotic pathway and activating the Akt/Nrf2/HO-1 pathway. J. Asian Nat. Prod. Res. 2013, 15, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Wang, S.; Duan, J.; Jia, N.; Zhu, Y.; Ding, Y.; Guan, Y.; Wei, G.; Yin, Y.; Xi, M.; et al. Protocatechualdehyde Protects Against Cerebral Ischemia-Reperfusion-Induced Oxidative Injury Via Protein Kinase Cε/Nrf2/HO-1 Pathway. Mol. Neurobiol. 2017, 54, 833–845. [Google Scholar] [CrossRef]
- Han, J.; Xiao, Q.; Lin, Y.; Zheng, Z.; He, Z.; Hu, J.; Chen, L. Neuroprotective effects of salidroside on focal cerebral ischemia/reperfusion injury involves the nuclear erythroid 2-related factor 2 pathway. Neural Regen. Res. 2015, 10, 1989. [Google Scholar] [CrossRef]
- Tang, C.; Hong, J.; Hu, C.; Huang, C.; Gao, J.; Huang, J.; Wang, D.; Geng, Q.; Dong, Y. Palmatine Protects against Cerebral Ischemia/Reperfusion Injury by Activation of the AMPK/Nrf2 Pathway. Oxid. Med. Cell. Longev. 2021, 2021, 1–12. [Google Scholar] [CrossRef]
- Fu, K.; Chen, M.; Zheng, H.; Li, C.; Yang, F.; Niu, Q. Pelargonidin ameliorates MCAO-induced cerebral ischemia/reperfusion injury in rats by the action on the Nrf2/HO-1 pathway. Transl. Neurosci. 2021, 12, 20–31. [Google Scholar] [CrossRef]
- Wu, G.; Zhu, L.; Yuan, X.; Chen, H.; Xiong, R.; Zhang, S.; Cheng, H.; Shen, Y.; An, H.; Li, T.; et al. Britanin Ameliorates Cerebral Ischemia–Reperfusion Injury by Inducing the Nrf2 Protective Pathway. Antioxid. Redox Signal. 2017, 27, 754–768. [Google Scholar] [CrossRef]
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic Cascades in Ischemic Stroke. Int. J. Stroke 2012, 7, 378–385. [Google Scholar] [CrossRef]
- Shaw, P.; Chattopadhyay, A. Nrf2–ARE signaling in cellular protection: Mechanism of action and the regulatory mechanisms. J. Cell. Physiol. 2020, 235, 3119–3130. [Google Scholar] [CrossRef]
- Stefanson, A.; Bakovic, M. Dietary Regulation of Keap1/Nrf2/ARE Pathway: Focus on Plant-Derived Compounds and Trace Minerals. Nutrients 2014, 6, 3777–3801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phipps, M.S.; Cronin, C.A. Management of acute ischemic stroke. BMJ 2020, 368, l6983. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Species (Sex; Age) | Experimental Model | #Findings Indicating Endogenous Modulation of Nrf2 after Ischemic Stroke | Tissue | Ref. | ||
|---|---|---|---|---|---|---|
| Specific Findings | General Effect | |||||
| MICE | ICR mice (M; 8 weeks) | MCAO/R (1 h/2, 8, 24, 72 h) | ↑ Nrf2, HO-1 and Trx protein expression ↓ Keap1 protein expression | ↑ | ischemic brain tissue | [90] |
| ddY mice (M; 8–12 weeks) | MCAO/R (1 h/6, 24, 48 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic cortex and striatum | [82] | |
| CD-1 mice (M; NI) | pMCAO (24 h) | ↑ nuclear Nrf2 translocation ↑ HO-1 protein expression ↑ SOD activity | ↑ | ischemic cerebral cortex | [91] | |
| ICR mice (M; 8–10 weeks) | MCAO/R (1 h/24 h) | ↑ Nrf2 and HO-1 mRNA levels ↑ nuclear Nrf2 and HO-1 protein expression | ↑ | ischemic cerebral cortex | [92] | |
| OKD48 mice (M, F; 11–13 weeks) | MCAO/R (45 min/ 12, 24, 72 h, 7 d) | ↑ Nrf2 and HO-1 protein expression | ↑ | peri-ischemic brain tissue | [93] | |
| Mice (NI) (M; 12 weeks) | MCAO/R (2 h/24 h) | ↑ Nrf2, HO-1, GCL protein expression | ↑ | ischemic brain tissue | [94] | |
| C57BL/6 mice (M; 8 weeks) | BCCAO/R (20 min/24 h) | ↑ Nrf2 DNA binding activity ↑ nuclear Nrf2, HO-1 and NQO1 protein expression | ↑ | striatum | [95] | |
| C57BL/6J mice (M; 8–10 weeks) | MCAO/R (1 h/24 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic brain tissue | [96] | |
| ICR mice (M; 6 weeks) | MCAO/R (1 h/24–72 h, 7 d) | ↑ Nrf2 protein expression ↓ Keap1 protein expression | ↑ | ischemic brain tissue | [97] | |
| C57BL/6 mice (M; 10–18 weeks) | pMCAO (1 d–3 d) | ↑ NQO1, HO-1, SOD2 and GPX-1 protein expression | ↑ | ischemic brain tissue | [89] | |
| C57BL/6 mice (M; 10–12 weeks) | MCAO/R (1.5 h/7 d) | ↑ Nrf2 and SOD1 protein expression | ↑ | ischemic brain tissue | [98] | |
| ddY mice (M; 5–8 weeks) | MCAO/R (2 h/2, 6, 22 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic brain tissue | [99] | |
| CD-1 mice (M; 4 weeks) | MCAO/R (1 h/24 h) | ↑ Nrf2 and HO-1 mRNA ↑ Nrf2 and HO-1 protein ↑ HO-1 activity | ↑ | ischemic brain tissue | [100] | |
| C57BL/6 mice (M; 12 weeks) | MCAO/R (1 h/3 d) | ↑ nuclear Nrf2, HO-1 and NQO1 protein expression | ↑ | cerebral tissue | [101] | |
| ICR mice (M; NI) | MCAO/R (2 h/24 h) | ↑ cytosolic Nrf2, HO-1 and NQO1 protein expression | ↑ | hippocampus | [102] | |
| BALB/c mice (M; 7 weeks) | MCAO/R (1.5 h/24–72 h) | ↑ Nrf2, HO-1 and iNOS protein expression ↓ SOD activity | ↑ | ischemic brain tissue | [103] | |
| ICR mice (M; 8 weeks) | p dMCAO (7 d) | ↑ Nrf2 protein expression | ↑ | ischemic cerebral cortex | [104] | |
| C57BL/6J mice (M; NI) | MCAO/R (1.5 h/24 h) | ↓ SOD 2, HO-1, NQO1 and Nrf2 protein expression ↓ SOD activity ↑ NADPH oxidase protein expression | ↓ | ischemic brain tissue | [105] | |
| C57BL/6 mice (M; NI) | MCAO/R (1 h/72 h) | ↓ Nrf2 protein expression | ↓ | hippocampus and cerebral cortex | [106] | |
| RATS | SD rats (M; adult) | MCAO/R (2 h/2, 6, 24 h) | ↑ Nrf2 and HO-1 protein expression ↑ Nrf2 and HO-1 mRNA levels | ↑ | ischemic cerebral cortex | [107] |
| SD rats (M; NI) | pMCAO (72 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic cerebral cortex | [108] | |
| SD rats (M; adult) | pMCAO (72 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic brain tissue | [109] | |
| SD rats (M; NI) | MCAO/R (70min/ 4, 24, 72 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic brain tissue | [110] | |
| SD rats (M; 9 weeks) | MCAO/R (1 h/24 h) | ↑ Nrf2 and NQO1 protein expression ↑ Nrf2 binding activity to ARE | ↑ | ischemic brain tissue | [111] | |
| SD rats (M; adult) | MCAO/R (2 h/22 h) | ↑ Nrf2 (nuclear) and HO-1 protein expression | ↑ | cerebral cortex | [112] | |
| SD rats (M; adult) | MCAO/R (2 h/24 h) | ↑ Nrf2 protein expression ↑ HO-1 protein expression | ↑ | ischemic brain tissue | [113] | |
| SD rats (M; NI) | pMCAO (24 h) | ↑ nuclear Nrf2 translocation ↑ HO-1 protein expression ↑ Nrf2 and HO-1 mRNA levels | ↑ | ischemic brain tissue | [114] | |
| SD rats (M; NI) | pMCAO (24 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic brain tissue | [115] | |
| SD rats (M; adult) | MCAO/R (1 h/ 72 h) | ↑ Nrf2 and NQO1 protein expression | ↑ | ischemic cerebral cortex | [116] | |
| Wistar rats (M; NI) | MCAO/R (1 h/72 h) | ↑ Nrf2, HO-1 and NQO1 mRNA levels | ↑ | ischemic brain tissue | [117] | |
| Wistar rats (M; 6 months) | BCCAO/R (30 min/72 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | hippocampus | [118] | |
| SD rats (M; NI) | pMCAO (72 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic brain tissue | [119] | |
| SD rats (M; 10–12 weeks) | MCAO/R (1 h/24 h) | ↑ Nrf2, Txr-1, Prdx1, Prdx2, Prdx3 and Prdx4 protein expression ↑ Nrf2, Txr-1, Prdx1, Prdx2, Prdx3 and Prdx4 mRNA levels | ↑ | ischemic brain tissue | [120] | |
| SD rats (M; NI) | MCAO/R (2 h/24 h) | ↑ Nrf2 protein expression ↑ Nrf2 mRNA levels ↓ SOD activity | ↑ | ischemic brain tissue | [121] | |
| SD rats (NI; NI) | BCCAO/R (10 min/1–7 d) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic brain tissue | [122] | |
| SD rats (M; adult) | MCAO/R (1.5 h/24 h) | ↑ Nrf2 and HO-1 protein expression ↑ Nrf2 and HO-1 mRNA levels | ↑ | hippocampus | [123] | |
| SD rats (M; adult) | pMCAO (24 h) | ↑ nuclear Nrf2 translocation ↑ HO-1 and SOD1 protein expression ↑ HO-1 mRNA levels ↑ SOD1 activity | ↑ | ischemic cerebral cortex | [124] | |
| SD rats (M; adult) | MCAO/R (2 h/ 72 h) | ↑ Nrf2 protein expression ↑ HO-1 protein expression | ↑ | ischemic brain tissue | [125] | |
| SD rats (M; NI) | MCAO/R (2 h/7 d) | ↑ Nrf2 protein expression | ↑ | ischemic brain tissue | [126] | |
| SD rats (M; adult) | MCAO/R (1 h/24 h) | ↑ Nrf2, NQO1 and Srnx1 protein expression ↑ Prdx 1, Prdx 2, Prdx 3, Prdx 4 protein expression | ↑ | ischemic brain tissue | [127] | |
| Hannover-Wistar rats (M; NI) | MCAO/R (1 h/24 h) | ↑ Nrf2 protein expression ↑ SOD and GPx activity | ↑ | hippocampus | [128] | |
| Wistar rats (M; adult) | BCCAO/R (45 min/24 h) | ↑ iNOS and Nrf2 protein expression | ↑ | hippocampus | [129] | |
| SD rats (M; 60–80 days) | MCAO/R (1 h/24 h) | ↑ Nrf2 and Trx1 mRNA levels ↑ Trx1 protein expression | ↑ | ischemic brain tissue | [130] | |
| SD rats (F; adult) | MCAO/R (1.5 h/72 h) | ↑ Nrf2 and NQO1 protein expression | ↑ | ischemic brain tissue | [131] | |
| Wistar rats (M; adult) | MCAO/R (1.5 h/72 h) | ↑ Nrf2, HO-1 and NQO1 mRNA levels | ↑ | ischemic brain tissue | [132] | |
| SD rats (M; adult) | PCI (20 min) | ↑ Nrf2 and HO-1 protein expression | ↑ | cerebral cortex ischemic penumbra | [133] | |
| SD rats (M; NI) | Focal PTI | ↑ Nrf2 and HO-1 protein expression | ↑ | penumbra of cerebral infarction | [134] | |
| SD rats (M; 7–10 weeks) | MCAO/R (2 h/72 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | cerebral cortex and striatum | [135] | |
| SD rats (M; NI) | MCAO/R (1 h/6 or 24 h) | ↑ Nrf2 protein expression ↓ SOD activity | ↑ | ischemic penumbra | [136] | |
| SD rats (M; Adult) | MCAO/R (2 h/72 h) | ↑ nuclear Nrf2 protein expression ↓ cytosolic Nrf2, NQO1 and HO-1 protein expression ↓ SOD activity | ↑ | ischemic brain tissue | [137] | |
| SD rats (M; 8 weeks) | MCAO/R (2 h/24 h) | ↑ HO-1 protein expression ↓ Nrf2 and Trx protein expression | ↑↓ | cerebral cortex | [113] | |
| SD rats (M; adult) | MCAO/R (2 h/72 h) | ↓ SOD activity ↓ Nrf2, HO-1 and NQO1 protein expression | ↓ | ipsilateral ischemic tissue | [138] | |
| SD rats (M; 10 months) | MCAO/R (2 h/48 h) | ↓ HO-1 protein expression ↓ SOD activity | ↓ | ipsilateral ischemic tissue | [139] | |
| Wistar rats (M; NI) | MCAO/R (2 h/24 h) | ↓ GPx and SOD activity ↓ Nrf2 and NQO1 protein expression | ↓ | ischemic brain tissue | [140] | |
| SD rats (M; 3 months) | MCAO/R (1.5 h/72 h) | ↓ NQO1, HO-1 and cytoplasmic Nrf2 protein expression | ↓ | ischemic brain tissue | [141] | |
| SD rats (M; 7–8 weeks) | MCAO/R (1.5 h/7 d) | ↓ Nrf2 mRNA levels | ↓ | ischemic brain tissue | [142] | |
| SD rats (M; NI) | MCAO/R (2 h/7 d) | ↓ SOD activity ↓ nuclear Nrf2, HO-1 and NQO1 protein expression ↓ Nrf2, HO-1 and NQO1 mRNA levels | ↓ | cerebral cortex | [143] | |
| SD rats (NI; NI) | BCCAO/R (10 min occlusion + 10 min reperfusion + 10 min occlusion) | ↓ Nrf2, NQO1 and HO-1 protein expression ↓ SOD activity | ↓ | ischemic brain tissue | [144] | |
| SD rats (F; NI) | MCAO/R (1 h/24 h) | ↓ Nrf2 protein expression | ↓ | hippocampus | [145] | |
| SD rats (M; NI) | MCAO/R (1.5 h/14 d) | ↓ Nrf2 protein expression ↑ Keap-1 protein expression ↓ SOD and catalase activities | ↓ | ischemic brain tissue | [146] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farina, M.; Vieira, L.E.; Buttari, B.; Profumo, E.; Saso, L. The Nrf2 Pathway in Ischemic Stroke: A Review. Molecules 2021, 26, 5001. https://doi.org/10.3390/molecules26165001
Farina M, Vieira LE, Buttari B, Profumo E, Saso L. The Nrf2 Pathway in Ischemic Stroke: A Review. Molecules. 2021; 26(16):5001. https://doi.org/10.3390/molecules26165001
Chicago/Turabian StyleFarina, Marcelo, Leonardo Eugênio Vieira, Brigitta Buttari, Elisabetta Profumo, and Luciano Saso. 2021. "The Nrf2 Pathway in Ischemic Stroke: A Review" Molecules 26, no. 16: 5001. https://doi.org/10.3390/molecules26165001
APA StyleFarina, M., Vieira, L. E., Buttari, B., Profumo, E., & Saso, L. (2021). The Nrf2 Pathway in Ischemic Stroke: A Review. Molecules, 26(16), 5001. https://doi.org/10.3390/molecules26165001