
Redox Conversions of 5-Methyl-6-nitro-7-oxo-4,7-dihydro-1,2,4triazolo[1,5-a]pyrimidinide L-Arginine Monohydrate as a Promising Antiviral Drug

 , , ,
, , ,  and
and
Abstract
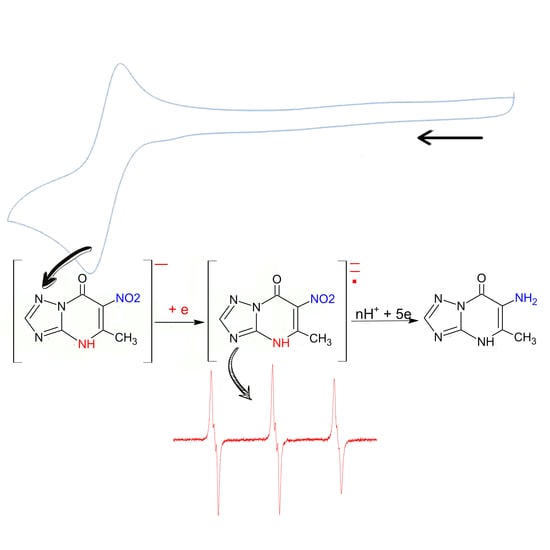
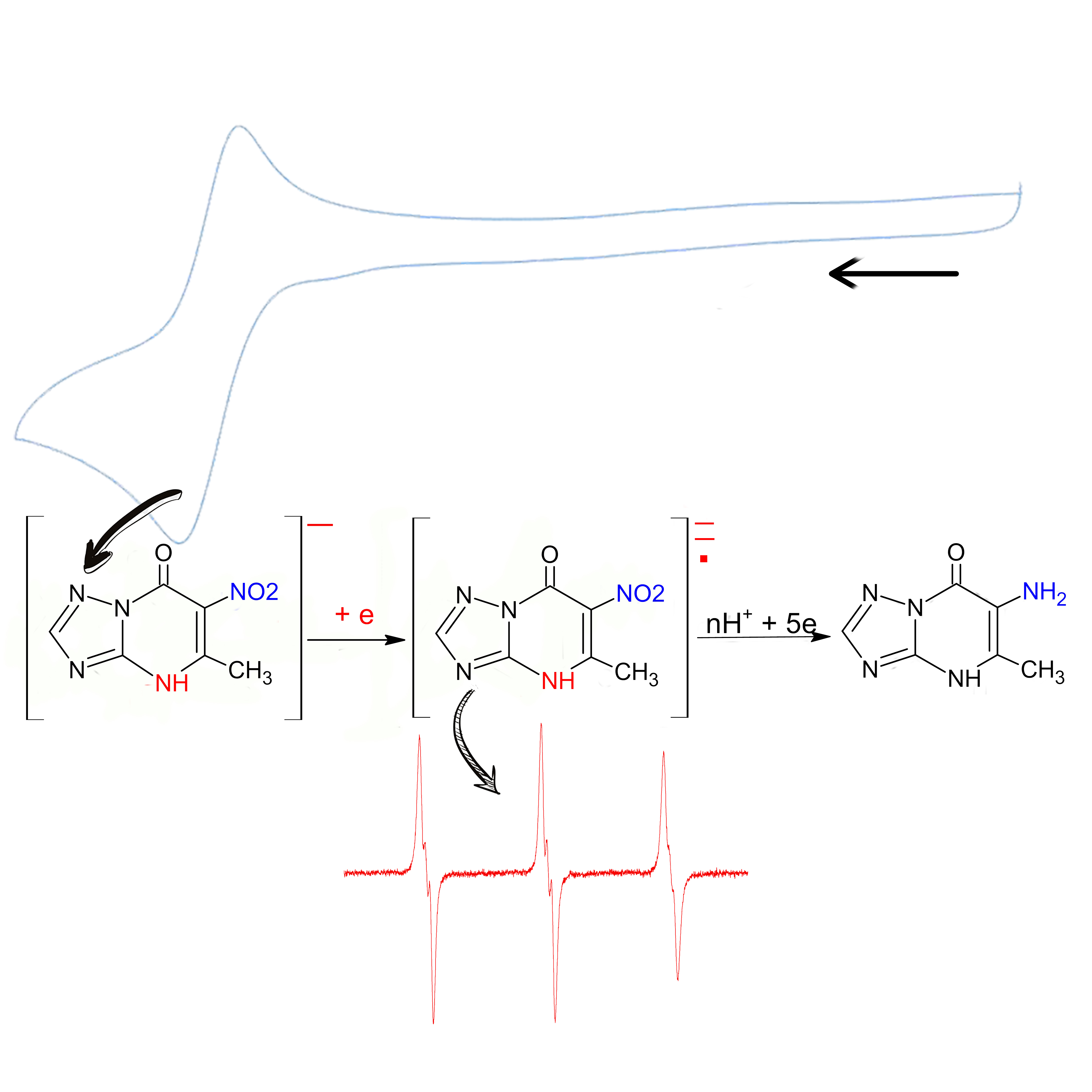
:
1. Introduction
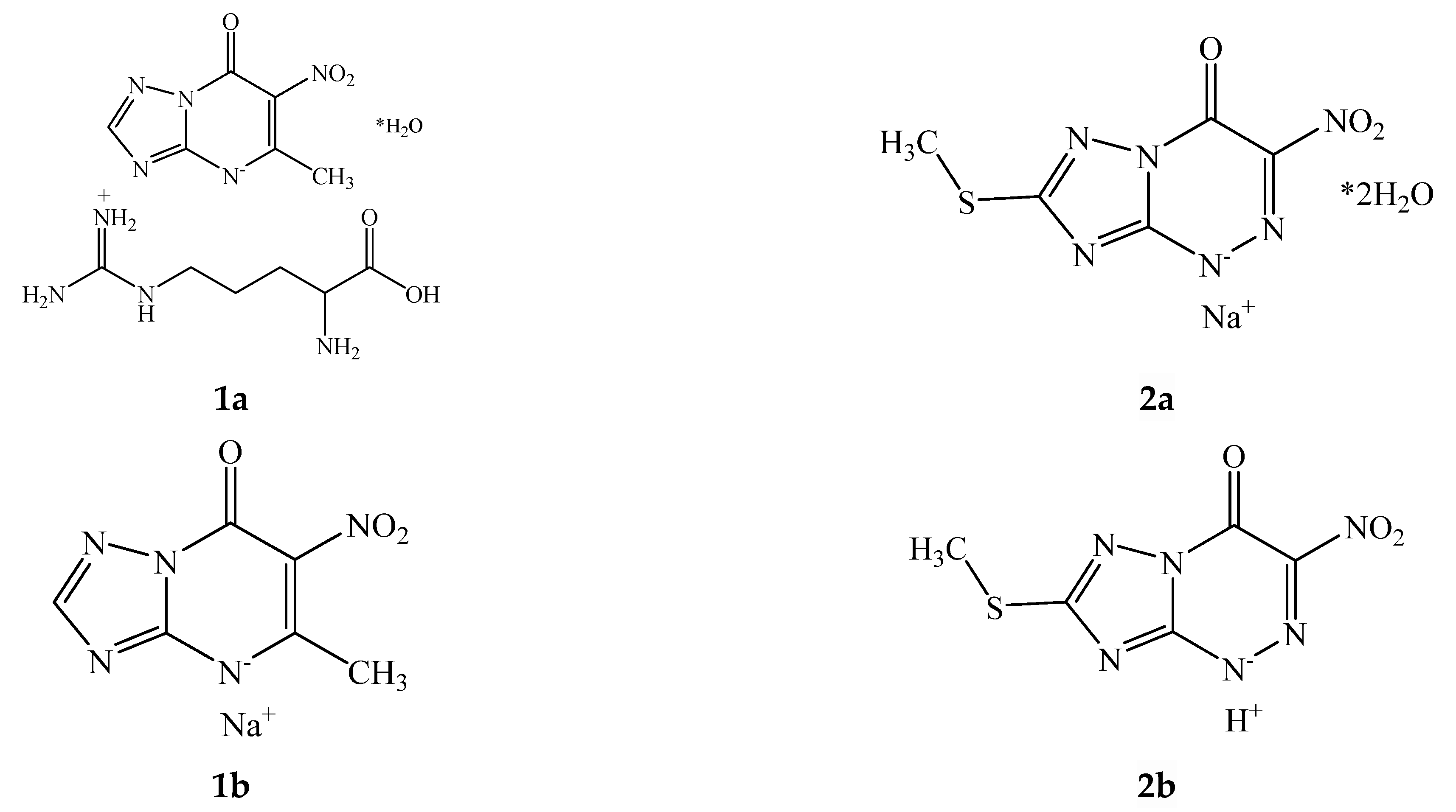
2. Experimental Section
2.1. Materials and Reagents
2.2. Electrochemical Devices and Methods
2.3. ESR Spectroscopy with Preliminary Electrochemical Generation
3. Results and Discussion
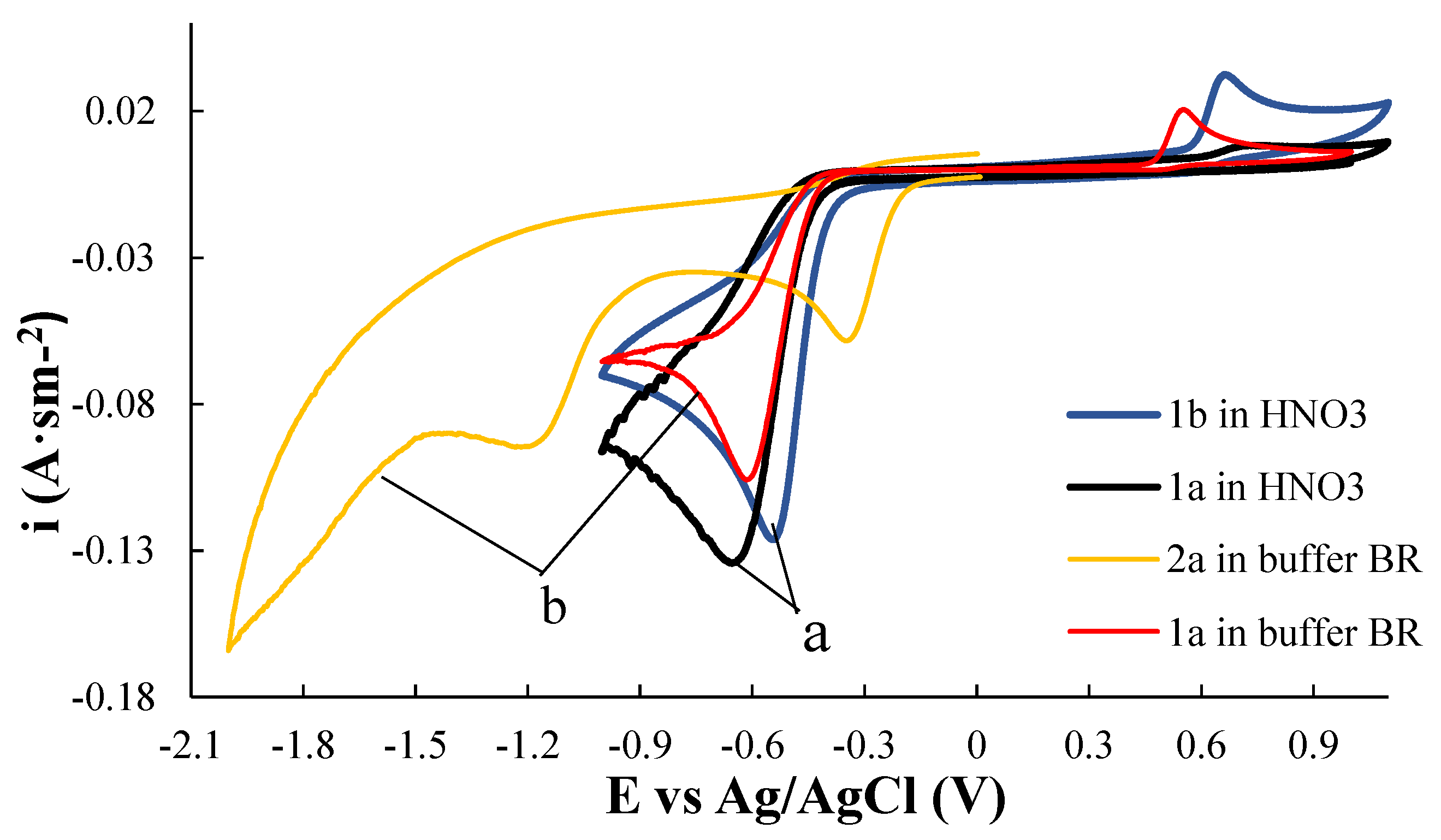
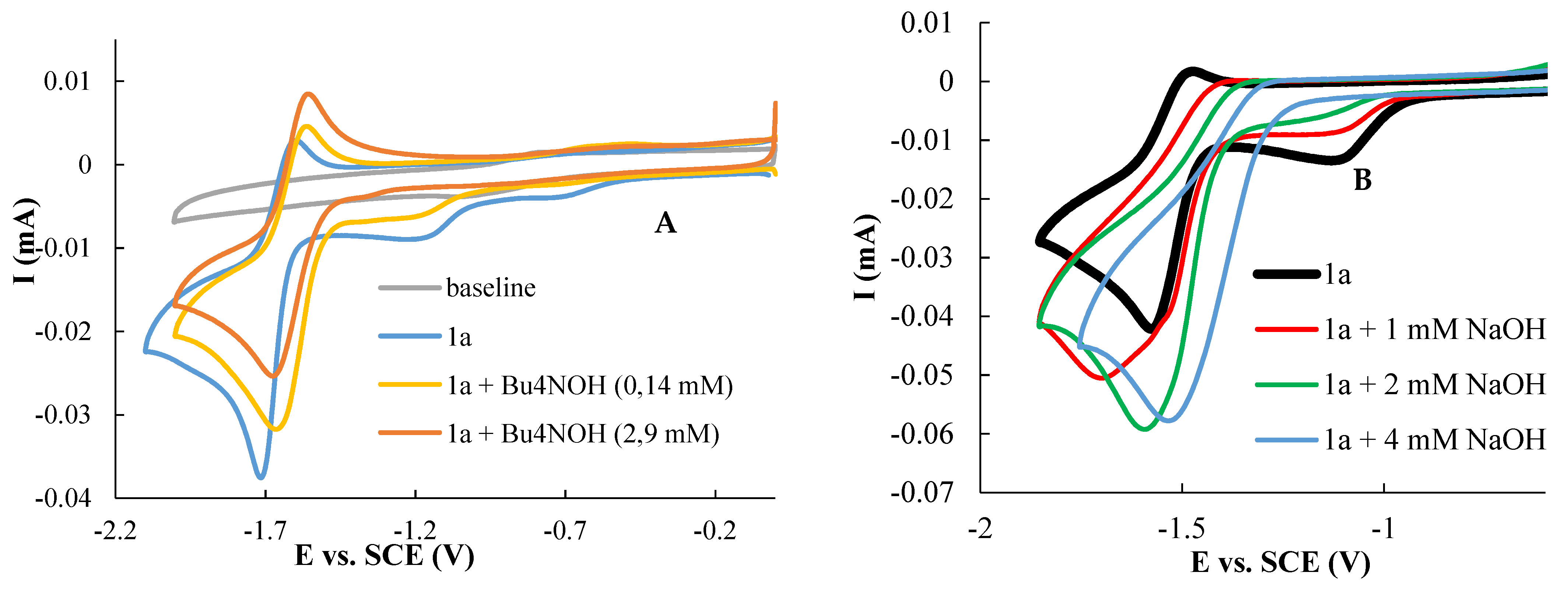
3.1. Electrochemical Behavior of Compounds 1a and 1b in Aqueous Media
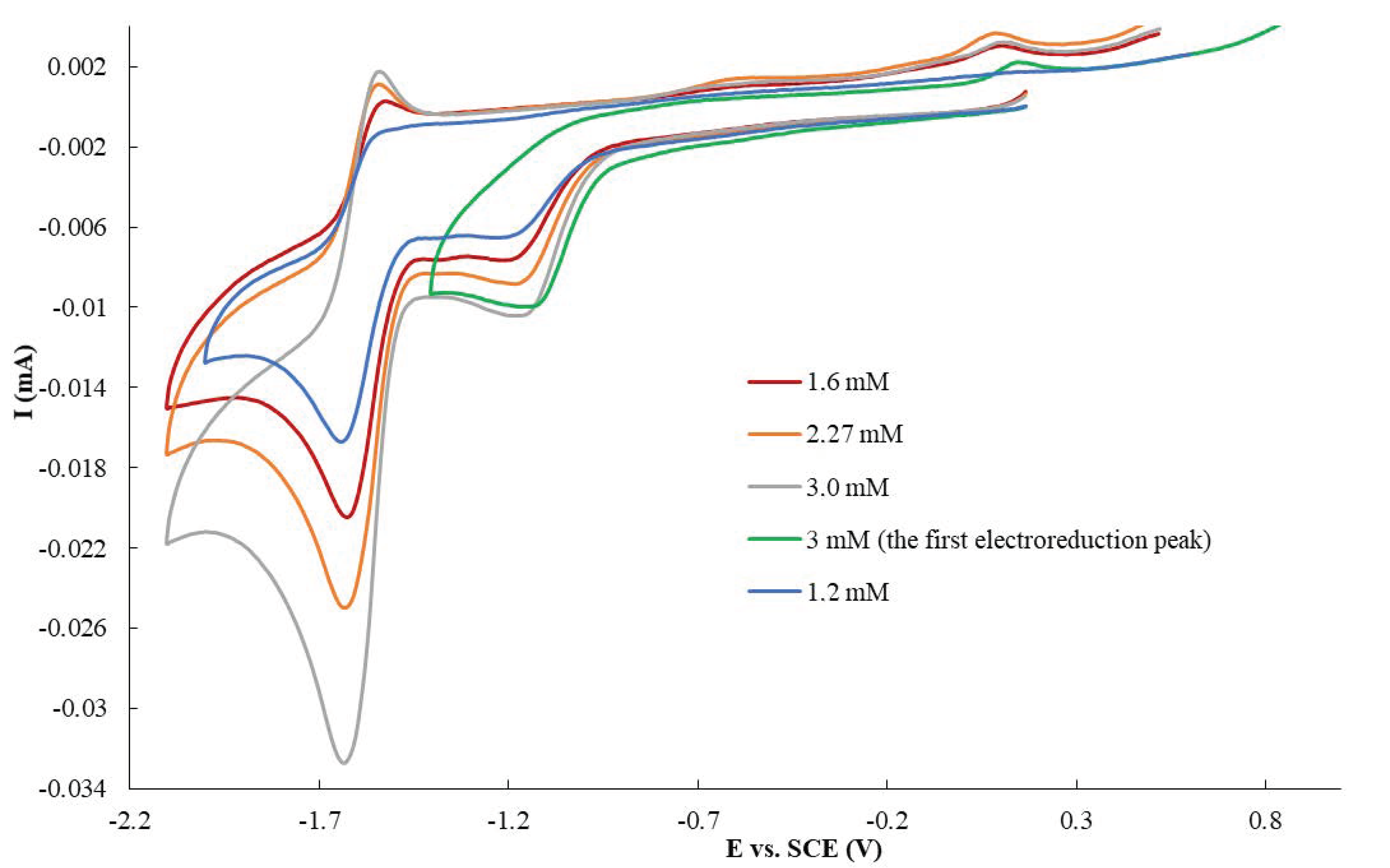
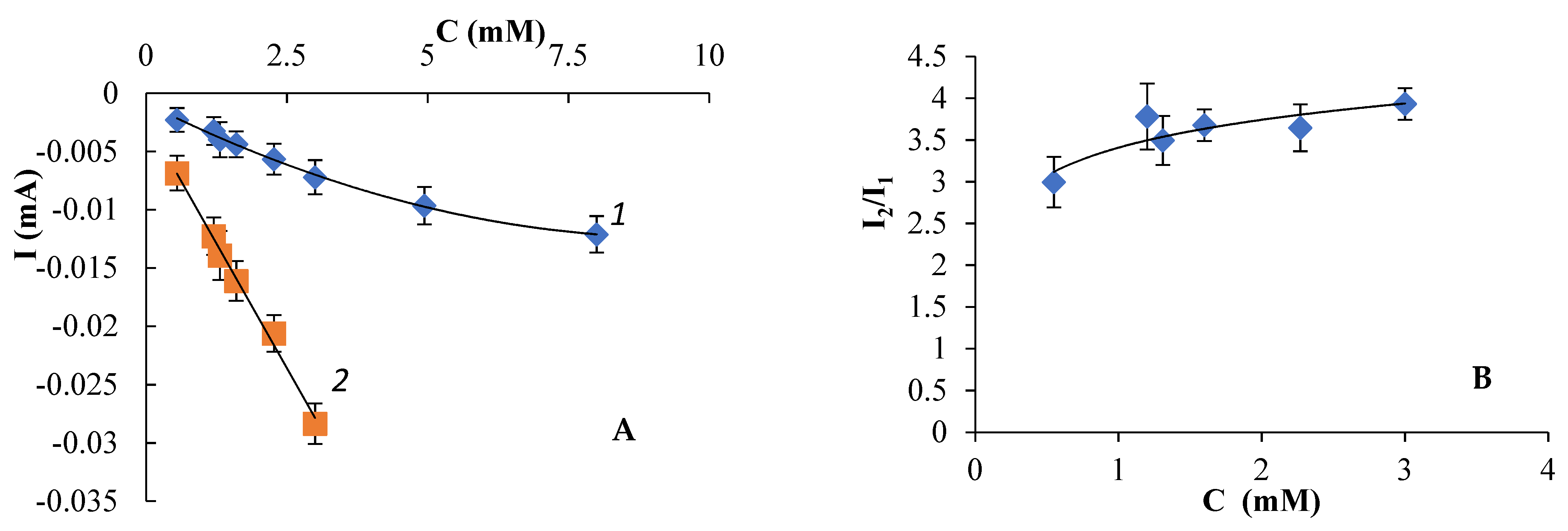
3.2. Electroreduction of Compound 1a in Aprotic Media
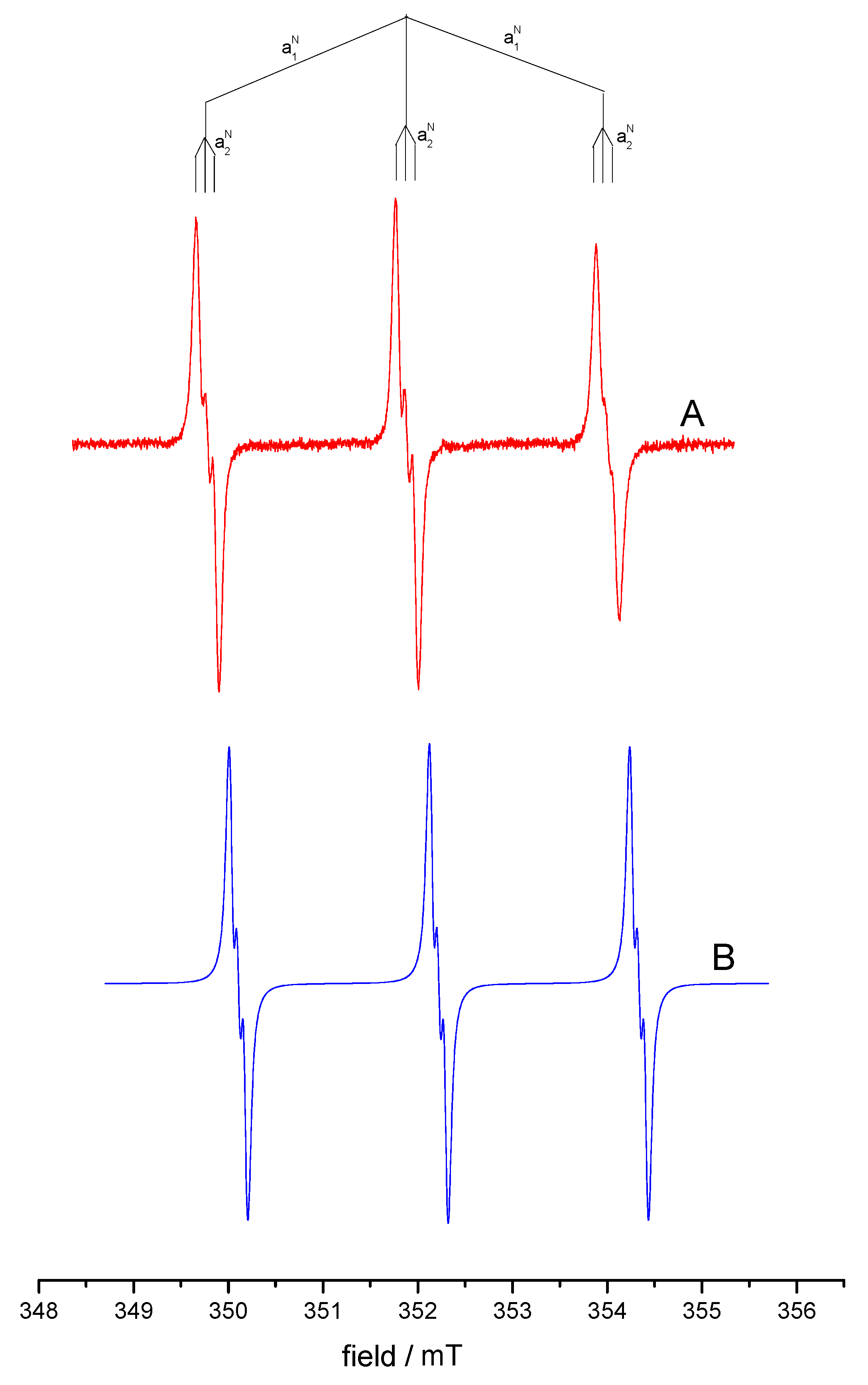
3.3. Study of the Products of Electroreduction of Compound 1a by ESR Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Nepali, K.; Lee, H.Y.; Liou, J.P. Nitro-Group-Containing Drugs. J. Med. Chem. 2018, 62, 2851–2893. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.F. Thiazolides: A new class of antiviral drugs. Expert Opin. Drug Metab. Toxicol. 2009, 5, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Otsuka and Mylan Announce License Agreement to Commercialize Delamanid (Deltyba™) for Multidrug-Resistant Tuberculosis (MDR-TB) in HighBurden Countries. PR Newswire: News distribution, targeting and monitoring. Available online: https://www.prnewswire.com/news-releases/otsuka-and-mylan-announce-license-agreement-to-commercialize-delamanid-deltyba-for-multidrug-resistant-tuberculosis-mdr-tb-in-high-burden-countries-300508333.html (accessed on 24 June 2021).
- Halsey, J.L. Current Approaches to the Treatment of Gastrointestial Infections: Focus on Nitazoxanide. Clin. Med. Ther. 2009, 1, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Kedderis, G.L.; Miwa, G.T. The metabolic activation of nitroheterocyclic therapeutic agents. Drug Metab. Rev. 1988, 19, 33–62. [Google Scholar] [CrossRef] [PubMed]
- Wardman, P. Application of pulse radiolysis methods to study the reactions and structure of biomolecules. Rep. Prog. Phys. 1978, 41, 259–296. [Google Scholar] [CrossRef]
- Grunberg, E.; Titsworth, E.H. Chemotherapeutic properties of heterocyclic compounds: Monocyclic compounds with five-membered rings. Annu. Rev. Microbiol. 1973, 27, 317–346. [Google Scholar] [CrossRef]
- Robins, R.K.; Revankar, G.R.; O’Brien, D.E.; Springer, R.H.; Albert, T.N.A.; Senga, K.; Miller, J.P.; Streeter, D.G. Purine analog inhibitors of xanthine oxidase-structure activity relationships and proposed binding of the molybdenum cofactor. J. Heterocycl. Chem. 1985, 22, 601–634. [Google Scholar] [CrossRef]
- Rusinov, V.L.; Charushin, V.N.; Chupakhin, O.N. Biologically Active Azolo-1,2,4-Triazines and Azolopyrimidines. Russ. Chem. Bull. 2018, 1, 573–599. [Google Scholar] [CrossRef]
- Sologub, T.V.; Tokin, I.I.; Midikari, A.S.; Tsvetkov, V.V. A comparative efficacy and safety of using antiviral drugs in therapy of patients with influenza. Infektsionnye Bolezn. 2017, 15, 25–32. [Google Scholar] [CrossRef]
- Lioznov, D.A.; Tokin, I.I.; Zubkova, T.G.; Sorokin, P.V.; Zhdanov. Practice of application of domestic anti-viral drug in etiotropic therapy of acute respiratory viral infection. Ter. Arkhiv 2021, 92, 59–63. [Google Scholar] [CrossRef]
- Tihonova, Е.P.; Yu, T.; Kuzmina, А.А.; Anisimova, Y.; Kalinina, S. Nitrosynthones in the synthesis of nitrodividuals of azolo [1,5-a] pyrimidines and azolo [5,1-c] -1,2,4-triazines. Eksperimentalnaya klynicheskaya Farmacol. 2018, 350, 25–32. [Google Scholar] [CrossRef]
- Kasyanenko, K.V.; Kozlov, K.V.; Maltsev, O.V.; Lapikov, I.I.; Gordienko, V.V.; Sharabhanov, V.V.; Sorokin, P.V.; Zhdanov, K.V. Evaluation of the effectiveness of Riamilovir in the complex therapy of patients with COVID-19. Ter. Arkhiv 2021, 93, 291–295. [Google Scholar] [CrossRef]
- Sabitov, A.U.; Sorokin, P.V.; Dashutina, S.Y. Experience of the preventive use of the drug Riamilovir in the foci of coronavirus infection (COVID-19). Ter. Arkhiv 2021, 93, 435–439. [Google Scholar] [CrossRef]
- Deyeva, E.G.; Shevchik, Y.I.; Shaldghan, A.A.; Zagorodnikova, K.A.; Tumashov, A.A.; Baklykov, A.V.; Kotovskaya, S.K.; Chupahin, O.N.; Charushin, V.N.; Rusinov, V.L.; et al. New antiviral drug TRIAZID. Results of first phase of clinical trial. Razrab. Regist. Lek. Sredstv 2018, 3, 172–180. [Google Scholar]
- Slepukhin, P.А.; Voinkov, E.K.; Ulomskiy, E.N.; Savateev, K.V.; Kopchuk, D.S.; Zyryanov, G.V.; Fedotov, V.V.; Charushin, V.N.; Chupakhin, O.N.; Rusinov, V.L. Synthesis and X-ray structural studies of 5-methyl-6-nitro-7-oxo-4,7-dihydro-1,2,4-triazolo[1,5-а]pyrimidine L-arginine and piperidine salts. Chem. Heterocycl. Compd. 2019, 55, 989–992. [Google Scholar] [CrossRef]
- Baizer, M.M.; Lund, H. Organic Electrochemistry; Lund, H., Ed.; Marcel Dekker: New York, NY, USA, 1983; ISBN 978-042-913-168-4. [Google Scholar]
- Kornilov, M.Y.; Turov, A.V.; Myasnikov, A.V.; Torgashev, P.A.; Rusinov, V.L.; Chupakhin, O.N. Aromaticity of nitroazoloazines with a bridging nitrogen atom. Zhurnal Org. Khimii. 1991, 27, 144–148. [Google Scholar]
- Zuman, P. Half a century of research using polarography. Microchem. J. 1997, 57, 4–51. [Google Scholar] [CrossRef]
- Ivoilova, A.V.; Mikhal’chenko, L.V.; Tsmokalyuk, A.N.; Kozitsina, A.N.; Ivanova, A.V.; Rusinov, V.L. Redox conversions of new antiviral drug Triazavirin®: Electrochemical study and ESR spectroscopy. Russ. Chem. Bull. 2021, 70, 1099–1108. [Google Scholar] [CrossRef]
- Malakhova, N.A.; Ivoilova, A.V.; Zamana, A.A.; Rusinov, V.L.; Alyamovskaya, I.S.; Ivanova, A.V.; Kozitsina, A.N. Quantitative Determination of the Main Substance of the Triazid® Antiviral Drug by Voltammetry. J. Anal. Chem. 2020, 75, 396–401. [Google Scholar] [CrossRef]
- Bull, O.N.; Chupakhin, V.N.; Charushin, V.L.; Rusinov, E.N.; Ulomsky, S.K.; Kotovskaya, O.I.; Kiselev, E.G.; Deeva, K.V.; Sa-vateev, S.S. Byul. Izobret 5-methyl-6-nitro-7-oxo-4,7-dihydro-1,2,4-triazolo [1,5-alpha] pyrimidinide l-argininium mono-hydrate. Borisov Patent RF 2529487, 20 October 2014. (In Russian). [Google Scholar]
- Bull Kiselev, O.I.; Petrov, A.Y.; Rusinov, V.L.; Ulomskiy, E.N.; Charushin, V.N.; Chupakhin, O.N. Izobret 5-methyl-6-nitro-1,2,4,-triazolo[1,5-a]pyrimidin-7-one dihydrate sodium salt. Patent RF 2330036, 27 July 2008. (In Russian). [Google Scholar]
- Bull, O.N.; Chupakhin, O.N.; Chupakhin, V.L.; Rusinov, E.N.; Ulomsky, E.N.; Ulomsky, V.N.; Charushin, V.N.; Charushin, A.Y.; Petrov, A.Y.; Petrov, O.I. IzobretSodium salt of 2-methylthio-6-nitro-1,2-4-triazolo[5,1-c]-1,2,4-triazin-7 (4h) -one, dihydrate with antiviral activity. Kiselev Patent RF 2294936, 10 March 2007. (In Russian). [Google Scholar]
- Lurie, Y.Y. Handbook of Analytical Chemistry; Chemistry: Moscow, Russia, 1971; ISBN 978-5-903034-26-0. [Google Scholar]
- Wilfred, L.F. Purification of Laboratory Chemicals; Perrin, D.D., Ed.; Elsevier: Amsterdam, The Netherlands, 2013; ISBN 0-08-034714-2. [Google Scholar]
- Dikalov, S.; Skatchkov, M.; Bassenge, E. Spin Trapping of Superoxide Radicals and Peroxynitriteby 1-Hydroxy-3-carboxy-pyrrolidine and 1-Hydroxy-2,2,6,6-tetramethyl-4-oxo-piperidine and the Stability ofCorresponding Nitroxyl Radicals Towards BiologicalReductants. Biochem. Biophys. Res. Commun. 1997, 231, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Kirilyuk, I.A.; Voinov, M.; Grigor’ev, I.A. ESR detection of cellular and mitochondrial superoxide using cyclic hydroxylamines. Free Radic. Res. 2011, 45, 417–430. [Google Scholar] [CrossRef]
- De Bono, D.; Yang, W.D.; Symons, M.C. The effect of myoglobin on the stability of the hydroxyl-radical adducts of 5, 5 dimethyl-1-pyrolline-N-oxide (DMPO), 3, 3, 5, 5 tetramethyl-1-pyrolline-N-oxide (TEMPO) and 1-alpha-phenyl-tert-butyl nitrone (PBN) in the presence of hydrogen peroxide. Free. Radic. Res. 1994, 20, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Migita, C.T.; Migita, K. Spin trapping of the nitrogen-centered radicals. Characterization of the DMPO/DEPMPO spin adducts. Chem. Lett. 2003, 32, 466–467. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 8, 1–6. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Avogadro: An Open-Source Molecular Builder and Visualization Tool, version 1.2; Available online: http://avogadro.cc/ (accessed on 2 May 2021).
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in ESR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Squella, J.A.; Bollo, S.; Núñez-Vergara, L.J. Recent Developments in the Electrochemistry of Some NitroCompounds of Biological Significance. Curr. Org. Chem. 2005, 9, 565–581. [Google Scholar] [CrossRef]
- Grimshaw, J. Electrochemical Reactions and Mechanisms in Organic Chemistry; Grimshaw, J., Ed.; Elsevier: Amsterdam, The Netherlands, 2000; ISBN 0-444-72007-3. [Google Scholar]
- Scholz, F.; Bond, A.M.; Compton, R.G.; Fiedler, D.A.; Inzelt, G.; Kahlert, H.; Komorsky-Lovric, Š.; Lohse, H.; Lovric, M.; Marken, F.; et al. Electroanalytical Methods. Guide to Experiments and Application; Scholz, F., Ed.; Springer: Berlin, Germany, 2002; ISBN 978-3-642-02915-8. [Google Scholar]
- Zuman, P.; Fijalek, Z.; Dumanovic, D.; Sužnjević, D. Polarographic and electrochemical studies of some aromatic and heterocyclic nitro compounds, part I: General mechanistic aspects. Electroanalysis 1992, 4, 783–794. [Google Scholar] [CrossRef]
- Karakus, C.; Zuman, P.J.; Karakus, C.; Zuman, P. Polarographic and electrochemical studies of some aromatic and heterocyclic nitro compounds Part 9. Substitutent effects on protonation of the radical ArNO2H and its reactions with hydroxylamino and nitroso compounds in buffered mixtures of water and DMF. Electroanal. Chem. 1995, 396, 499–505. [Google Scholar] [CrossRef]
- Rusakov, A.I.; Mendkovich, A.S.; Gultyay, V.P.; Orlov, V.Y. Structure and Reactivity of Organic Radical Anions; Mir: Moscow, Russia, 2005; ISBN 5-03-003772-1. [Google Scholar]
- Michelson, A.M. Ions and Ion Pairs in Organic Reactions; Szwarc, M., Ed.; John Wiley and Sons: New York, NY, USA, 1972. [Google Scholar]
- Cavalcanti, J.C.; Oliveira, N.V.; de Moura, M.A.B.; Fruttero, R.; Bertinaria, M.; Goulart, M.O. Evidence of self-protonation on the electrodic reduction mechanism of an anti-Helicobacter pylorimetronidazole isotere. Electroanal. Chem. 2004, 571, 177–182. [Google Scholar] [CrossRef]
- Maldonado, T.; Martínez-González, E.; Frontana, C. Intramolecular Hydrogen Bonding/Self protonation Processes Modulated by the Substituent Effect in Hydroxyl-substituted Naphthoquinones. Electroanalysis 2016, 28, 2827–2833. [Google Scholar] [CrossRef]
- Jakubke, H.-D.; Eshkite, H. Amino Acids, Peptides, Proteins; Mitin, Y.V., Ed.; Mir: Moscow, Russia, 1985; ISBN 180-300-000-017-1. [Google Scholar]
- Lopyrev, V.; Larina, L.; Rakhmatulina, T.; Shibanova, E.; Vakulskaia, T.; Voronkov, M. Dianion radicals of nitro derivatives of pyrazole, imidazole and 1,2,4-triazole. Fiz. Khimiya 1978, 242, 142–145. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
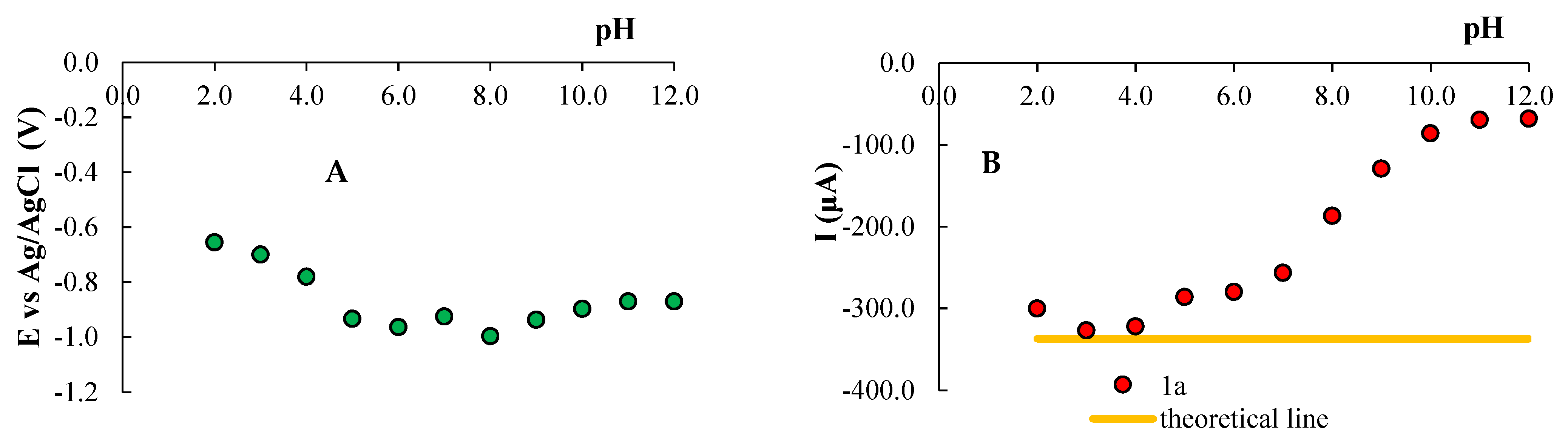{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ne | ne (CA) | ne (CC) | ne (RDE) |
|---|---|---|---|---|
| 1a | 5.63 | 5.72 | 5.86 | 4.59 |
| K3Fe(CN)6 | 1 | 1.02 | 1 | 0.96 |
| 2a [20] | 3.25 | 4.08 | 4.17 | 3.68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivoilova, A.; Mikhalchenko, L.V.; Tsmokalyuk, A.; Leonova, M.; Lalov, A.; Mozharovskaia, P.; Kozitsina, A.N.; Ivanova, A.V.; Rusinov, V.L. Redox Conversions of 5-Methyl-6-nitro-7-oxo-4,7-dihydro-1,2,4triazolo[1,5-a]pyrimidinide L-Arginine Monohydrate as a Promising Antiviral Drug. Molecules 2021, 26, 5087. https://doi.org/10.3390/molecules26165087
Ivoilova A, Mikhalchenko LV, Tsmokalyuk A, Leonova M, Lalov A, Mozharovskaia P, Kozitsina AN, Ivanova AV, Rusinov VL. Redox Conversions of 5-Methyl-6-nitro-7-oxo-4,7-dihydro-1,2,4triazolo[1,5-a]pyrimidinide L-Arginine Monohydrate as a Promising Antiviral Drug. Molecules. 2021; 26(16):5087. https://doi.org/10.3390/molecules26165087
Chicago/Turabian StyleIvoilova, Alexandra, Ludmila V. Mikhalchenko, Anton Tsmokalyuk, Marina Leonova, Andrey Lalov, Polina Mozharovskaia, Alisa N. Kozitsina, Alla V. Ivanova, and Vladimir L. Rusinov. 2021. "Redox Conversions of 5-Methyl-6-nitro-7-oxo-4,7-dihydro-1,2,4triazolo[1,5-a]pyrimidinide L-Arginine Monohydrate as a Promising Antiviral Drug" Molecules 26, no. 16: 5087. https://doi.org/10.3390/molecules26165087
APA StyleIvoilova, A., Mikhalchenko, L. V., Tsmokalyuk, A., Leonova, M., Lalov, A., Mozharovskaia, P., Kozitsina, A. N., Ivanova, A. V., & Rusinov, V. L. (2021). Redox Conversions of 5-Methyl-6-nitro-7-oxo-4,7-dihydro-1,2,4triazolo[1,5-a]pyrimidinide L-Arginine Monohydrate as a Promising Antiviral Drug. Molecules, 26(16), 5087. https://doi.org/10.3390/molecules26165087